Mutagénese
A mutagénese é o processo pelo qual a informação genética de um organismo muda devido à origem de uma mutação. Pode ocorrer espontaneamente na natureza ou como resultado da exposição a mutagénios. Pode ser conseguida experimentalmente usando procedimentos laboratoriais. Um mutagénio é um agente causador de mutações (agente mutagénico), que pode ser químico ou físico, que resulta num aumento na taxa de mutação na informação genética de um organismo. Na natureza, a mutagénese pode conduzir à formação de um cancro e a diversas doenças hereditárias, mas é também uma força que impulsiona a evolução. A mutagénese como ciência desenvolveu-se baseando-se nos trabalhos que fizeram Hermann Muller, Charlotte Auerbach e J. M. Robson na primeira metade do século XX.[1]
História
O ADN pode ser modificado de forma natural ou artificialmente por diversos agentes físicos, químicos e biológicos, originando mutações. Hermann Muller descobriu no início da década de 1920 que as "altas temperaturas" têm a capacidade de fazer mutar genes,[2] e em 1927 demonstrou a existência de uma ligação causal da irradiação com raios X com a mutação, experimentando com uma máquina de raios X, notando alterações genéticas quando irradiava moscas do vinagre com uma dose relativamente alta.[3][4] Muller observou vários rearranjos cromossómicos neste experimento e sugeriu que as mutações podem ser causa do cancro.[5][6] A associação da exposição à radiação com o cancro tinha sido observada já em 1902, seis anos depois da descoberta dos raios X por Wilhelm Röntgen, e a descoberta da radioatividade por Henri Becquerel.[7] Lewis Stadler, contemporâneo de Muller, também mostrou o efeito dos raios X sobre as mutações na cevada em 1928, e da radiação ultravioleta (UV) no milho em 1936.[8] Na década de 1940, Charlotte Auerbach e J. M. Robson descobriram que o gás mostarda pode também causar mutações nas moscas do vinagre.[9] Embora as alterações nos cromossomas causadas pelos raios X e pelo gás mostarda fossem facilmente observáveis pelos primeiros investigadores, outras alterações no ADN induzidas por outros mutagénios não eram tão fáceis de observar; o mecanismo pelo qual ocorrem pode ser complexo e levar muito tempo a ser descoberto. Por exemplo, foi sugerido que a fuligem causa cancro já em 1775,[10] e foi demonstrado que o alcatrão de hulha causa também cancro em 1915.[11] Os compostos químicos envolvidos em ambos os casos foram depois identificados como hidrocarbonetos aromáticos policíclicos (HAPs).[12] Os HAPs por si sós não são carcinogénicos e foi proposto em 1950 que as formas carcinogénicas dos HAPs são os óxidos produzidos como metabolitos nos processos celulares.[13] O processo metabólico identificou-se na década de 1960 como uma catálise do citocromo P450, que produz espécies reativas, que podem interagir com o ADN para formar adutos, ou moléculas produto resultantes da reação;[14][15] o mecanismo pelo qual os adutos HAPs dão origem a mutações está ainda a ser investigado.
Distinção entre mutação e danos no ADN
Os danos no ADN são alterações anormais na estrutura do ADN que não podem, por si sós, ser replicados quando ocorre a replicação do ADN. Em contraste, uma mutação é uma alteração na sequência de um ácido nucleico que pode ser replicada; portanto, uma mutação pode ser herdada de uma geração para a seguinte. Os danos podem ocorrer pela adição de um agente químico (aduto), ou pela alteração estrutural de uma base do ADN (criando um nucleótido anormal ou fragmento de nucleótido) ou uma quebra numa das fitas do ADN ou em ambas. Tais danos no ADN podem ter como resultado uma mutação. Quando um ADN que contém danos é replicado, pode ser inserida uma base incorreta na nova fita complementar à medida que é sintetizada (ver reparação do ADN § Síntese translesão). A inserção incorreta na nova fita ocorre em frente do local danificado na fita molde e esta inserção incorreta pode converter-se numa mutação (isto é, um par de bases mudado) na próxima ronda de replicação. Além disso, as quebras de dupla fita no ADN podem ser reparadas por um processo de reparação impreciso, como é a união de extremos não homólogos, que produz mutações. As mutações podem normalmente ser evitadas se sistemas precisos de reparação do ADN reconhecerem o dano no ADN e o repararem antes que a próxima ronda de replicação se complete. São empregues direta ou indiretamente pelo menos 169 enzimas na reparação do ADN ou influenciam os processos de reparação do ADN. Destes, 83 são empregues diretamente nos cinco tipos de processos de reparação (ver reparação do ADN). O ADN nuclear de mamífero pode sofrer mais de 60.000 eventos de danos por célula e dia (ver danos no ADN (por causas naturais)). Se não forem corrigidos, estes adutos, depois de uma replicação incorreta dos locais danificados, pode originar uma mutação. Na natureza, as mutações que se originam podem ser benéficas ou deletérias e isto é uma força que impulsiona a evolução. Um organismo pode adquirir novas características por meio de mutações genéticas, mas as mutações podem também ter como resultado a alteração ou perda de função de um gene e, em casos graves, causar a morte do organismo. A mutação é também uma fonte principal para a aquisição de resistência a antibióticos em bactérias, e a agentes antifúngicos em leveduras e bolores.[16][17] Num laboratório, a mutagénese é uma técnica útil para gerar mutações que permitam examinar a função dos genes e produtos génicos em detalhe, produzindo proteínas ou cepas com funções e propriedades úteis melhoradas ou novas. Inicialmente a capacidade das radiações e os mutagénios químicos de causar mutações foi aproveitada para gerar mutações ao acaso, mas técnicas posteriores permitiram criar mutações específicas. Nos humanos, são transmitidas em média 60 novas mutações de progenitores a descendentes. Os varões tendem a passar mais mutações dependendo da sua idade, transmitindo em média duas novas mutações à sua progénie por cada ano adicional de idade.[18][19]
Mecanismos
A mutagénese pode ser de origem endógena (por exemplo, hidrólise espontânea), por meio de processos celulares que podem gerar espécies reativas do oxigénio e adutos do ADN ou por erros na replicação e reparação do ADN.[20] A mutagénese pode também ocorrer como resultado da presença de mutagénios ambientais que induzem alterações no ADN de um organismo. O mecanismo pelo qual ocorre a mutação varia de acordo com o mutagénio ou o agente causal envolvido. A maioria dos mutagénios atua direta ou indiretamente por meio de metabolitos mutagénicos sobre o ADN de um organismo, produzindo lesões. No entanto, alguns mutagénios podem afetar a replicação ou o mecanismo de repartição cromossómica e outros processos celulares. A mutagénese pode também ser autoinduzida por organismos unicelulares quando as condições ambientais são restritivas para o crescimento do organismo, como o crescimento bacteriano na presença de antibióticos, o crescimento de leveduras na presença de agentes antifúngicos ou o crescimento de outros organismos unicelulares num ambiente que carece de um nutriente essencial.[21][22][23] Muitos mutagénios químicos necessitam de ativação biológica para serem mutagénicos. Um grupo importante de enzimas que intervêm na geração de metabolitos mutagénicos é o citocromo P450.[24] Outras enzimas que podem também produzir metabolitos mutagénicos são a glutatião S-transferase e a epóxido hidrolase microssomal. Os mutagénios que não são mutagénicos em si, mas que necessitam de ativação biológica, denominam-se promutagénios. Embora a maioria dos mutagénios produza efeitos que finalmente resultam em erros na replicação, por exemplo, criando adutos que interferem com a replicação, alguns mutagénios podem afetar diretamente o processo de replicação ou reduzir a sua fidelidade. Análogos de bases como o 5-bromouracilo podem substituir a timina na replicação. Os metais como o cádmio, crómio e níquel podem aumentar a mutagénese de diversas maneiras além do dano direto ao ADN, por exemplo, reduzindo a capacidade de reparar erros, assim como produzindo alterações epigenéticas.[25] As mutações originam-se como resultado de problemas causados por lesões do ADN durante a replicação, o que resulta em erros na replicação. Nas bactérias, os grandes danos no ADN devido a mutagénios causam lacunas numa das fitas do ADN durante a replicação. Isto induz a resposta SOS, um processo de reparação de emergência que é também propenso ao erro, gerando assim mutações. Em células de mamíferos, a paragem da replicação nos locais danificados induz vários mecanismos de resgate que ajudam a contornar a zona das lesões no ADN; no entanto, isto pode também causar erros. A família Y das ADN polimerases é especializada em contornar a zona da lesão no ADN por um processo denominado síntese translesão, no qual estas polimerases de bypass da lesão substituem as ADN polimerases replicativas de alta fidelidade que ficaram paradas, transitando pela lesão e alongando o ADN até que se passa da zona da lesão para que a replicação normal possa recomeçar; estes processos podem ser propensos ao erro ou livres de erro.
Danos no ADN e mutação espontânea
O número de eventos de danos no ADN que ocorrem numa célula de mamíferos por dia é alto (mais de 60.000 por dia) e representa um problema para os organismos vivos que deve ser minimizado. A maioria das mutações espontâneas origina-se geralmente por síntese translesão propensa ao erro ao passar por um local com danos na fita molde durante a replicação do ADN. Este processo pode potencialmente superar os bloqueios letais no ADN, mas ao custo de introduzir imprecisões no ADN-filho. As relações causais dos danos no ADN com a mutação espontânea são ilustradas pelo crescimento aeróbico da bactéria Escherichia coli, na qual 89% das mutações de substituição de bases que ocorrem espontaneamente são causadas por efeito de espécies reativas do oxigénio.[26] Nas leveduras, mais de 60% de substituições de um único par de bases espontâneas e as deleções são provavelmente causadas por síntese translesão.[27] Uma fonte adicional significativa de mutações em eucariotas é a imprecisão do processo de reparação do ADN da união de extremos não homólogos, que é empregue frequentemente na reparação de quebras de dupla fita.[28] Em geral, parece que a principal causa subjacente de mutações espontâneas é a síntese translesão propensa ao erro durante a replicação do ADN e que a reparação por união de extremos não homólogos pode ser um importante contribuinte em eucariotas.
Hidrólise espontânea
O ADN não é completamente estável em solução aquosa e pode ocorrer a despurinação do ADN. Em condições fisiológicas, a ligação glicosídica pode ser hidrolisada espontaneamente e estima-se que 10.000 locais de purina no ADN são despurinados cada dia numa célula.[20] Existem inúmeras vias de reparação do ADN; no entanto, se o local apurínico não for reparado, pode ocorrer uma incorporação incorreta de nucleótidos durante a replicação. A adenina é incorporada preferencialmente pelas ADN polimerases num local apurínico. A citidina pode também ser desaminada a uridina a uma taxa que é 5% da taxa de despurinação e pode originar uma transição de G para A. As células eucariotas podem também conter 5-metilcitosina, que se crê estar envolvida no controlo da transcrição génica, a qual pode ser desaminada a timina.
Tautomeria
A tautomerização é o processo pelo qual os compostos se rearranjam espontaneamente para adotar as suas formas de isómero estrutural. Por exemplo, as formas ceto (C=O) da guanina e timina podem rearranjar-se nas suas raras formas enol (-OH), enquanto que as formas amino (-NH2) da adenina e citosina podem originar as mais raras formas imino (=NH). Na replicação do ADN, a tautomerização altera os locais de emparelhamento de bases e pode causar o emparelhamento impróprio das bases dos ácidos nucleicos.[29]
Modificação de bases
As bases dos ácidos nucleicos podem ser modificadas endogenamente por moléculas celulares normais. Por exemplo, o ADN pode ser metilado pela S-adenosilmetionina, que assim altera a expressão do gene marcado pela metilação sem causar nenhuma mutação na sequência do ADN. A modificação de histonas é um processo relacionado no qual as proteínas histonas à volta das quais o ADN se enrola podem ser também modificadas por metilação, fosforilação, ou acetilação; estas modificações podem atuar para alterar a expressão génica do ADN local e podem também atuar para sinalizar localizações onde há ADN danificado que se deve reparar. O ADN pode também ser glicosilado por açúcares redutores. Todas estas alterações são de tipo epigenético, já que não afetam a sequência de bases. Muitos compostos como os hidrocarbonetos aromáticos policíclicos (HAPs), aminas aromáticas, aflatoxina e alcaloides de pirrolizidina, podem formar espécies reativas do oxigénio catalisadas pelo citocromo P450. Estes metabolitos formam adutos no ADN, os quais podem causar erros na replicação, e os volumosos adutos aromáticos podem formar intercalações estáveis entre as bases e bloquear a replicação. Os adutos podem também induzir alterações conformacionais no ADN. Alguns adutos podem além disso originar a despurinação do ADN;[30] no entanto, não se sabe a importância da despurinação causada pelos adutos na geração de mutações. A alquilação e arilação de bases podem causar erros na replicação. Alguns agentes alquilantes, como as N-nitrosaminas podem requerer a reação catalítica do citocromo P450 para a formação de um catião alquílico reativo. O N7 e o O6 da guanina e o N3 e o N7 da adenina são mais suscetíveis ao ataque. Os adutos no N7 da guanina formam a maioria dos adutos do ADN, mas parece que não são mutagénicos. No entanto, a alquilação em O6 da guanina é daninha porque a reparação por excisão do aduto em O6 da guanina pode ser deficiente nalguns tecidos como o do cérebro.[31] A metilação em O6 da guanina pode ter como resultado uma transição de G para A, enquanto que a metilamina em O4 pode ser emparelhada incorretamente com a guanina. O tipo de mutação gerada pode, não obstante, depender do tamanho e tipo de aduto assim como da sequência do ADN.[32] As radiações ionizantes e as espécies reativas do oxigénio oxidam frequentemente a guanina, produzindo 8-oxoguanina.
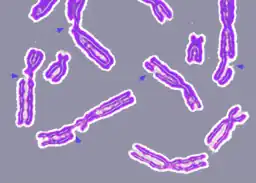
Danos no esqueleto molecular do ADN
A radiação ionizante pode produzir radicais livres muito reativos que podem quebrar as ligações no ADN. As quebras de dupla fita são especialmente daninhas e difíceis de reparar, produzindo translocações e deleções de partes de um cromossoma. Os agentes alquilantes como o gás mostarda podem também causar quebras no esqueleto do ADN. O stress oxidativo pode também gerar potentes espécies reativas do oxigénio que podem danificar o ADN. A reparação incorreta de outros danos induzidos pelas espécies do oxigénio altamente reativas pode originar mutações.
Ligações cruzadas
As ligações covalentes entre as bases dos nucleótidos do ADN da mesma fita ou de fitas opostas denominam-se ligações cruzadas no ADN; o estabelecimento de ligações cruzadas pode afetar tanto a replicação como a transcrição do ADN e pode ser causado pela exposição a diversos agentes. Alguns são compostos químicos que ocorrem na natureza e que também promovem as ligações cruzadas, como os psoralenos depois da ativação pela radiação UV e o ácido nitroso. As ligações cruzadas entre fitas opostas causam mais danos, já que bloqueiam a replicação e transcrição e podem causar quebras cromossómicas e rearranjos. Alguns compostos que facilitam as ligações cruzadas, como a ciclofosfamida, a mitomicina C e a cisplatina utilizam-se na quimioterapia anticancro devido ao seu alto grau de toxicidade para as células proliferantes.
Dimerização
A dimerização consiste na união de dois monómeros para formar um oligómero, como a formação de dímeros de pirimidinas como resultado da exposição a radiação UV, o que promove a formação de um anel ciclobutano entre timinas adjacentes no ADN.[33] Em células humanas, podem formar-se milhares de dímeros por dia devido à exposição normal à luz solar. A ADN polimerase η pode também ajudar a contornar estas lesões, mas de um modo propenso ao erro;[34] no entanto, os indivíduos com uma função de reparação do ADN defeituosa, como os que sofrem xeroderma pigmentosum, são sensíveis à luz do sol e têm mais risco de ter cancro de pele.
Clinicamente, pode distinguir-se se um tumor se formou como consequência direta da radiação UV por análise de sequenciação do ADN para o padrão de dimerização específico de contexto caraterístico que aparece devido à exposição excessiva ao sol.[35]
Intercalação entre bases
A estrutura planar de compostos químicos como o brometo de etídio e a proflavina permite-lhes inserirem-se entre as bases do ADN. Esta inserção faz com que o esqueleto da molécula de ADN se estique e aumenta a probabilidade de ocorrer o deslizamento (slippage) no ADN durante a replicação, já que as ligações entre as cadeias se tornam menos estáveis. O slippage para a frente originará uma mutação por deleção, enquanto que na direção inversa causará uma mutação por inserção. Além disso, a intercalação no ADN de antraciclinas como a daunorrubicina e a doxorrubicina interfere com o funcionamento da enzima topoisomerase II, bloqueando a replicação e causando recombinação homóloga mitótica.
Mutagénese insercional
Os transposãos e os vírus ou retrotransposões podem inserir sequências de ADN em regiões codificantes ou elementos funcionais de um gene e resultam na inativação do gene. Isto denomina-se mutagénese insercional.[36]
Mecanismos da mutagénese adaptativa
A mutagénese adaptativa compreende os mecanismos de mutagénese que permitem que um organismo se adapte a um stress ambiental. Como a variedade de stresses ambientais é muito ampla, os mecanismos que a permitem são também bastante diversos, como demonstram as investigações realizadas neste campo. Por exemplo, em bactérias, a modulação da resposta SOS e a síntese de ADN de prófago endógeno aumenta a resistência de Acinetobacter baumannii à ciprofloxacina.[16] Os mecanismos de resistência presumem-se estar ligados a mutações cromossómicas intransferíveis por transferência horizontal de genes nalguns membros da família Enterobacteriaceae, como Escherichia coli, Salmonella spp., Klebsiella spp. e Enterobacter spp.[37] Os eventos cromossómicos, especialmente a amplificação génica, parecem ser relevantes nesta mutagénese adaptativa em bactérias.[38] As investigações feitas em células eucariotas são muito mais escassas, mas os eventos cromossómicos parecem também ser bastante relevantes: enquanto foi relatado que a recombinação intracromossómica ectópica está envolvida na aquisição de resistência à 5-fluorocitosina em Saccharomyces cerevisiae,[17] descobriu-se que as duplicações genómicas lhe dão resistência a esta mesma espécie aos ambientes com escassez de nutrientes.[21][39][40]
Aplicações em laboratório
No laboratório, a mutagénese é uma técnica pela qual se geram deliberadamente por engenharia mutações no ADN para produzir genes mutantes, proteínas ou cepas de organismos. Podem ser mutados vários constituintes de um gene, como os seus elementos de controlo e os seus produtos génicos, o que permite que a função de um gene ou proteína possa ser examinada em detalhe. A mutação pode também produzir proteínas mutantes com propriedades alteradas interessantes ou melhorar ou criar uma nova função que possa ser útil comercialmente. Podem ser geradas também cepas mutantes de organismos que têm aplicações práticas ou que permitem investigar as bases moleculares de uma determinada função da célula. Os primeiros métodos de mutagénese produziam mutações completamente ao acaso; no entanto, os métodos modernos de mutagénese podem produzir mutações específicas de sítio. Entre as modernas técnicas de laboratório usadas para gerar estas mutações estão:
- Mutagénese sítio-dirigida / mutagénese de PCR
- Mutagénese insercional
- Mutagénese marcada com assinatura
- Mutagénese de transposão
- Mutagénese de saturação de sequências
Referências
- ↑ Beale, G. (1993). «The Discovery of Mustard Gas Mutagenesis by Auerbach and Robson in 1941». Genetics. 134 (2): 393–399. PMC 1205483
 . PMID 8325476. doi:10.1093/genetics/134.2.393
. PMID 8325476. doi:10.1093/genetics/134.2.393
- ↑ Kevin M. Gleason Published: 2017-03-07. «Hermann Joseph Muller's Study of X-rays as a Mutagen, (1926-1927)»
- ↑ «Genetics and Genomics Timeline 1927 Hermann J. Muller (1890-1967) demonstrates that X rays can induce mutations»
- ↑ Muller, H. J. (1927). «Artificial Transmutation of the Gene» (PDF). Science. 66 (1699): 84–87. Bibcode:1927Sci....66...84M. PMID 17802387. doi:10.1126/science.66.1699.84
- ↑ Crow, J. F.; Abrahamson, S. (1997). «Seventy Years Ago: Mutation Becomes Experimental». Genetics. 147 (4): 1491–1496. PMC 1208325. PMID 9409815. doi:10.1093/genetics/147.4.1491
- ↑ Calabrese, E. J. (30 junho de 2011). «Muller's Nobel lecture on dose–response for ionizing radiation:ideology or science?» (PDF). Archives of Toxicology. 85 (4): 1495–1498. PMID 21717110. doi:10.1007/s00204-011-0728-8. Consultado em 30 dezembro de 2011. Arquivado do original (PDF) em 2 de agosto de 2017
- ↑ Ronald L. Kathren (dezembro de 2002). «Historical Development of the Linear Nonthreshold Dose-Response Model as Applied to Radiation». University of New Hampshire Law Review. 1 (1)
- ↑ Stadler, L. J.; G. F. Sprague (15 de outubro de 1936). «Genetic Effects of Ultra-Violet Radiation in Maize. I. Unfiltered Radiation» (PDF). Proc. Natl. Acad. Sci. U.S.A. 22 (10): 572–8. Bibcode:1936PNAS...22..572S. PMC 1076819. PMID 16588111. doi:10.1073/pnas.22.10.572. Consultado em 11 de outubro de 2007
- ↑ Auerbach, C.; Robson, J.M.; Carr, J.G. (março de 1947). «Chemical Production of Mutations». Science. 105 (2723): 243–7. Bibcode:1947Sci...105..243A. PMID 17769478. doi:10.1126/science.105.2723.243
- ↑ Brown, J. R.; Thornton, J. L. (1957). «Percivall Pott (1714-1788) and Chimney Sweepers' Cancer of the Scrotum». British Journal of Industrial Medicine. 14 (1): 68–70. PMC 1037746. PMID 13396156. doi:10.1136/oem.14.1.68
- ↑ Yamagawa K, Ichikawa K (1915). «Experimentelle Studie ueber die Pathogenese der Epithel geschwuelste». Mitteilungen aus der Medizinischen Fakultät der Kaiserlichen Universität zu Tokyo. 15: 295–344
- ↑ Luch, Andreas (2005). «Nature and Nurture — Lessons from Chemical Carcinogenesis: Chemical Carcinogens — From Past to Present». Medscape
- ↑ Boyland E (1950). «The biological significance of metabolism of polycyclic compounds». Biochemical Society Symposium. 5: 40–54. ISSN 0067-8694. OCLC 216723160
- ↑ Omura, T.; Sato, R. (1962). «A new cytochrome in liver microsomes». The Journal of Biological Chemistry. 237 (4): 1375–1376. PMID 14482007. doi:10.1016/S0021-9258(18)60338-2
- ↑ Conney, A. H. (1982). «Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons: G. H. A. Clowes Memorial Lecture». Cancer Research. 42 (12): 4875–4917. PMID 6814745
- ↑ a b Geisinger, Edward; Vargas-Cuebas, Germán; Mortman, Nadav J.; Syal, Sapna; Dai, Yunfei; Wainwright, Elizabeth L.; Lazinski, David; Wood, Stephen; Zhu, Zeyu (11 de junho de 2019). Miller, Samuel I., ed. «The Landscape of Phenotypic and Transcriptional Responses to Ciprofloxacin in Acinetobacter baumannii : Acquired Resistance Alleles Modulate Drug-Induced SOS Response and Prophage Replication». mBio. 10 (3). ISSN 2150-7511. PMC 6561030. PMID 31186328. doi:10.1128/mBio.01127-19
- ↑ a b Quinto-Alemany, David; Canerina-Amaro, Ana; Hernández-Abad, Luís G.; Machín, Félix; Romesberg, Floyd E.; Gil-Lamaignere, Cristina (31 de julho de 2012). Sturtevant, Joy, ed. «Yeasts Acquire Resistance Secondary to Antifungal Drug Treatment by Adaptive Mutagenesis». PLOS ONE. 7 (7): e42279. Bibcode:2012PLoSO...742279Q. ISSN 1932-6203. PMC 3409178. PMID 22860105. doi:10.1371/journal.pone.0042279
- ↑ Jha, Alok (22 de agosto de 2012). «Older fathers pass on more genetic mutations, study shows». The Guardian
- ↑ Kong, A.; Frigge, M. L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S. A.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; Wong, W. S.; Sigurdsson, G.; Walters, G. B.; Steinberg, S.; Helgason, H.; Thorleifsson, G.; Gudbjartsson, D. F.; Helgason, A.; Magnusson, O. T.; Thorsteinsdottir, U.; Stefansson, K. (2012). «Rate of de novo mutations and the importance of father's age to disease risk». Nature. 488 (7412): 471–475. Bibcode:2012Natur.488..471K. PMC 3548427. PMID 22914163. doi:10.1038/nature11396
- ↑ a b Loeb, L. A. (1989). «Endogenous carcinogenesis: Molecular oncology into the twenty-first century--presidential address» (PDF). Cancer Research. 49 (20): 5489–5496. PMID 2676144
- ↑ a b Heidenreich, Erich (janeiro de 2007). «Adaptive Mutation in Saccharomyces cerevisiae». Critical Reviews in Biochemistry and Molecular Biology. 42 (4): 285–311. ISSN 1040-9238. PMID 17687670. doi:10.1080/10409230701507773
- ↑ Quinto-Alemany, David; Canerina-Amaro, Ana; Hernández-Abad, Luís G.; Machín, Félix; Romesberg, Floyd E.; Gil-Lamaignere, Cristina (31 de julho de 2012). Sturtevant, Joy, ed. «Yeasts Acquire Resistance Secondary to Antifungal Drug Treatment by Adaptive Mutagenesis». PLOS ONE. 7 (7): e42279. Bibcode:2012PLoSO...742279Q. ISSN 1932-6203. PMC 3409178. PMID 22860105. doi:10.1371/journal.pone.0042279
- ↑ Aghapour, Zahra; Gholizadeh, Pourya; Ganbarov, Khudaverdi; bialvaei, Abed Zahedi; Mahmood, Suhad Saad; Tanomand, Asghar; Yousefi, Mehdi; Asgharzadeh, Mohammad; Yousefi, Bahman (abril de 2019). «Molecular mechanisms related to colistin resistance in Enterobacteriaceae». Infection and Drug Resistance. 12: 965–975. PMC 6519339. PMID 31190901. doi:10.2147/idr.s199844
- ↑ Trevor M. Penning (2011). Chemical Carcinogenesis (Current Cancer Research). [S.l.]: Springer. ISBN 978-1617379949
- ↑ Salnikow K, Zhitkovich (janeiro de 2008). «Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium». Chemical Research in Toxicology. 21 (1): 28–44. PMC 2602826. PMID 17970581. doi:10.1021/tx700198a
- ↑ Sakai A, Nakanishi M, Yoshiyama K, Maki H (julho de 2006). «Impact of reactive oxygen species on spontaneous mutagenesis in Escherichia coli». Genes Cells. 11 (7): 767–78. PMID 16824196. doi:10.1111/j.1365-2443.2006.00982.x
- ↑ Kunz BA, Ramachandran K, Vonarx EJ (abril de 1998). «DNA sequence analysis of spontaneous mutagenesis in Saccharomyces cerevisiae». Genetics. 148 (4): 1491–505. PMC 1460101. PMID 9560369. doi:10.1093/genetics/148.4.1491
- ↑ Huertas P (janeiro de 2010). «DNA resection in eukaryotes: deciding how to fix the break». Nat. Struct. Mol. Biol. 17 (1): 11–6. PMC 2850169. PMID 20051983. doi:10.1038/nsmb.1710
- ↑ Sinden, Richard R. (1994). DNA Structure and Function. [S.l.]: Academic Press. pp. 17–20. ISBN 978-0126457506
- ↑ Melendez-Colon, V. J.; Smith, C. A.; Seidel, A.; Luch, A.; Platt, K. L.; Baird, W. M. (1997). «Formation of stable adducts and absence of depurinating DNA adducts in cells and DNA treated with the potent carcinogen dibenzoa, lpyrene or its diol epoxides». Proceedings of the National Academy of Sciences of the United States of America. 94 (25): 13542–13547. Bibcode:1997PNAS...9413542M. PMC 28342. PMID 9391062. doi:10.1073/pnas.94.25.13542
- ↑ Boysen, G.; Pachkowski, B. F.; Nakamura, J.; Swenberg, J. A. (2009). «The Formation and Biological Significance of N7-Guanine Adducts». Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 678 (2): 76–94. PMC 2739241. PMID 19465146. doi:10.1016/j.mrgentox.2009.05.006
- ↑ Loechler, E. L. (1996). «The role of adduct site-specific mutagenesis in understanding how carcinogen-DNA adducts cause mutations: Perspective, prospects and problems». Carcinogenesis. 17 (5): 895–902. PMID 8640935. doi:10.1093/carcin/17.5.895
- ↑ Setlow, R. B. (1966). «Cyclobutane-type pyrimidine dimers in polynucleotides». Science. 153 (734): 379–386. Bibcode:1966Sci...153..379S. PMID 5328566. doi:10.1126/science.153.3734.379
- ↑ Broyde, S.; Patel, D. J. (2010). «DNA repair: How to accurately bypass damage». Nature. 465 (7301): 1023–1024. Bibcode:2010Natur.465.1023B. PMC 4986998. PMID 20577203. doi:10.1038/4651023a
- ↑ Mata, Douglas A.; Williams, Erik A.; Sokol, Ethan; Oxnard, Geoffrey R.; Fleischmann, Zoe; Tse, Julie Y.; Decker, Brennan (23 março de 2022). «Prevalence of UV Mutational Signatures Among Cutaneous Primary Tumors». JAMA Network Open. 5 (3): e223833. PMID 35319765. doi:10.1001/jamanetworkopen.2022.3833
- ↑ Mohanasundaram, Boominathan; Rajmane, Vyankatesh B.; Jogdand, Sukanya V.; Bhide, Amey J.; Banerjee, Anjan K. (junho de 2019). «Agrobacterium-mediated Tnt1 mutagenesis of moss protonemal filaments and generation of stable mutants with impaired gametophyte». Molecular Genetics and Genomics. 294 (3): 583–596. PMID 30689096. doi:10.1007/s00438-019-01532-4
- ↑ Aghapour, Zahra; Gholizadeh, Pourya; Ganbarov, Khudaverdi; bialvaei, Abed Zahedi; Mahmood, Suhad Saad; Tanomand, Asghar; Yousefi, Mehdi; Asgharzadeh, Mohammad; Yousefi, Bahman (abril de 2019). «Molecular mechanisms related to colistin resistance in Enterobacteriaceae». Infection and Drug Resistance. 12: 965–975. PMC 6519339. PMID 31190901. doi:10.2147/idr.s199844
- ↑ Hersh, Megan N; Ponder, Rebecca G; Hastings, P.J; Rosenberg, Susan M (junho de 2004). «Adaptive mutation and amplification in Escherichia coli: two pathways of genome adaptation under stress». Research in Microbiology. 155 (5): 352–359. PMID 15207867. doi:10.1016/j.resmic.2004.01.020
- ↑ Longerich, S.; Galloway, A. M.; Harris, R. S.; Wong, C.; Rosenberg, S. M. (19 de dezembro de 1995). «Adaptive mutation sequences reproduced by mismatch repair deficiency.». Proceedings of the National Academy of Sciences. 92 (26): 12017–12020. Bibcode:1995PNAS...9212017L. ISSN 0027-8424. PMC 40287. PMID 8618835. doi:10.1073/pnas.92.26.12017
- ↑ Rosenberg, Susan M.; Fitzgerald, Devon M. (1 de abril de 2019). «What is mutation? A chapter in the series: How microbes "jeopardize" the modern synthesis». PLOS Genetics. 15 (4): e1007995. ISSN 1553-7404. PMC 6443146. PMID 30933985. doi:10.1371/journal.pgen.1007995