Glicogênio fosforilase
A Glicogénio Fosforilase é uma enzima fosforilase (EC 2.4.1.1) que catalisa a libertação de moléculas de glicose 1-fosfato a partir da molécula de glicogénio. Este é o passo limitante da glicogenólise nos animais. As glicoses são libertadas nas ligações glicosídicas terminais $\alpha$-1,4 das cadeias de glicogénio. A glicogénio fosforilase é também estudada como uma proteína modelo regulada por fosforilação reversível (ligação covalente reversível) e por efeitos alostéricos.
Mecanismo
A reação global catalisada é:
(cadeia de glicogênio α-1,4)n + Pi ↔ (cadeia de glicogênio α-1,4)n-1 + D-glicose 1-fosfato.[1]
A glicogênio fosforilase quebra o glicogênio em unidades de glicose, que são libertadas na forma de glicose 1-fosfato. O glicogênio fica cada vez que o enzima intervém com uma unidade de glicose a menos. Para que a glicose libertada possa ser utilizada no metabolismo deve converter-se em glicose 6-fosfato pelo enzima fosfoglicomutase.
Embora a reação seja reversível em solução, dentro da célula o enzima só funciona na direção degradativa porque a concentração de fosfato inorgânico (Pi) é muito maior que a de glicose 1-fosfato.[1]
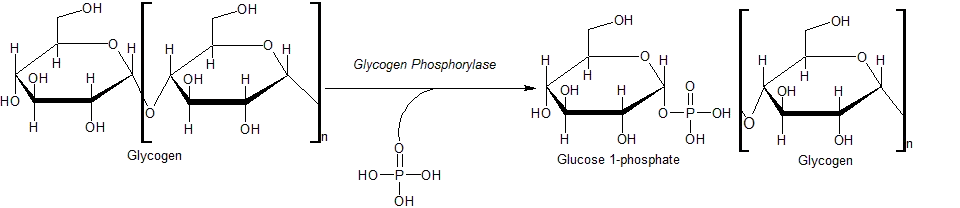
A glicogênio fosforilase pode atuar só sobre cadeias lineares de glicogênio (com ligação glicosídica α1-4). A sua atividade cessa imediatamente quatro resíduos antes de se chegar a uma ligação α1-6, que estão nos pontos de ramificação do glicogênio, muito comuns na molécula. Nesta situação, é necessário que atue outro enzima chamado enzima desramificador do glicogênio, o qual deixará reta a cadeia nessa zona, e pode realizar nos eucariotas duas atividades (em E. coli fazem-nas dois enzimas distintos): atividade transferase e atividade α-1,6-glicosidase. Primeiramente atua como enzima transferase e muda um bloco de três resíduos de glicose da rama externa para a extremidade de outra cadeia, e depois atua como α1-6 glicosidase e quebra o resíduo de glicose que fica com ligação α1-6 na nova cadeia linear. Depois de se ter feito tudo isto, a glicogênio fosforilase pode continuar a separar mais resíduos de glicose. O enzima é específico de cadeias α1-4, já que tem uma fenda de 30 ángstrom de comprimento com o mesmo raio que a hélice formada pela cadeia de glicogênio; na fenda cabem 4-5 resíduos de glicose, mas é demasiado estreita para as zonas com ramificações. Esta fenda conecta o sítio de armazenamento do glicogênio no enzima com o sítio ativo catalítico.
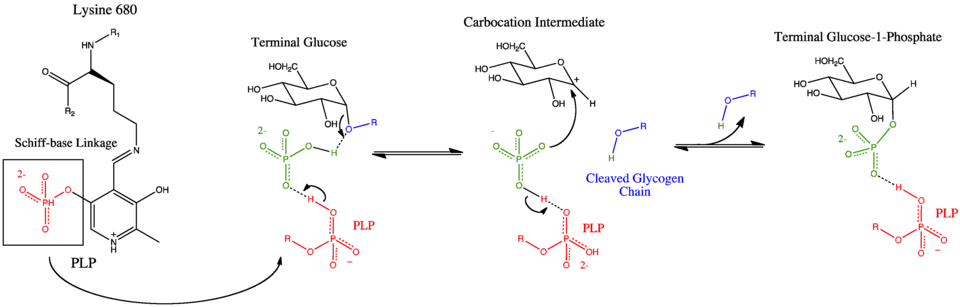
O mecanismo ilustra-se na figura de abaixo. A glicogênio fosforilase tem um fosfato de piridoxal (PLP, derivado da vitamina B6) em cada sítio catalítico ou ativo. O fosfato de piridoxal está ligado a resíduos básicos (neste caso lisina, a Lys680) e forma por ligação covalente uma base de Schiff. Uma vez que se forma a base de Schiff, mantendo a molécula de PLP no sítio ativo, o grupo fosfato do PLP (em vermelho na figura) facilmente doa um protão a uma molécula de fosfato inorgânico (em verde), o que faz com que o fosfato inorgânico seja por sua vez desprotonado pelo oxigénio da ligação glicosídica α-1,4 (em azul). A doada desprotonação do PLP deve-se a que a sua carga negativa não está só estabilizada no grupo fosfato, mas também no anel de piridina, e assim a base conjugada originada pela desprotonação do PLP é bastante estável. O oxigénio (azul) protonado agora é um bom grupo de saída lábil (leaving group), e a cadeia de glicogênio separa-se da glicose terminal numa reação SN1, originando a formação de uma molécula de glicose com um carbocatião secundário na posição 1. Finalmente, o fosfato inorgânico desprotonado atua como um nucleófilo e liga-se com o carbocatião, formando glicose 1-fosfato e uma cadeia de glicogênio com um resíduo de glicose a menos. Na reação liberta-se uma molécula de água, mas não é uma hidrólise, mas sim uma fosforólise.
Propôs-se também um mecanismo alternativo que implica a intervenção de um oxigénio carregado positivamente em conformação de meia-cadeira.[2]
Estrutura
O monómero da glicogênio fosforilase é uma proteína grande de 842 aminoácidos com uma massa de 97.434 Da nas células musculares. O enzima pode existir na sua forma inativa como monómero ou tetrâmero, mas a sua forma biologicamente ativa é um dímero formado por duas subunidades idênticas (homodímero).[3]
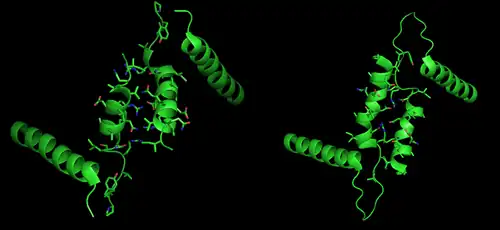
O dímero de glicogênio fosforilase tem muitas regiões de importância biológica, incluindo os sítios catalíticos, os sítios de ligação ao glicogênio ("de armazenamento"), os sítios alostéricos, e um resíduo de serina que pode ser fosforilado reversivelmente.
Os sítios catalíticos estão relativamente enterrados dentro da molécula, a 15 Å da superfície da proteína e da interface da subunidade.[4] Este difícil acesso ao sítio catalítico a partir da superfície é significativo no sentido de que faz com que a atividade da proteína seja muito suscetível à regulação, já que pequenos efeitos alostéricos poderiam aumentar muito o acesso relativo do glicogênio ao sítio.
Talvez o sítio regulatório mais importante seja Ser14, o sítio onde se produz a fosforilação reversível muito próximo da interface da subunidade. A mudança estrutural associada com a fosforilação, e com a conversão da fosforilase b em fosforilase a, é a disposição que adotam os resíduos 10 a 22 originalmente desordenados que passam a formar α hélices. Esta mudança aumenta a atividade fosforilase até num 25% mesmo na ausência de AMP, e além disso potencia a ativação feita pelo AMP.[5]
O sítio alostérico para o AMP nas isoformas musculares deste enzima está próximo à interface da subunidade, tal como a Ser14. Quando se liga um AMP a este sítio, o que corresponde a uma mudança do estado T (tenso) do enzima para o estado R (relaxado), origina pequenas mudanças na estrutura terciária na interface da subunidade que causam grandes mudanças na estrutura quaternária.[6] A ligação do AMP faz rodar as regiões da molécula chamadas hélices torre (resíduos 262-278) das duas subunidades 50˚ uma em relação à outra por meio de um aumento na organização e interações entre as subunidades. Esta rotação das hélices torre causa a rotação das duas subunidades cerca de 10˚ uma em relação à outra, e o que é mais importante, desordena os resíduos 282-286 (a alça 280s) que bloqueia o acesso ao sítio catalítico no estado T mas não no estado R.[4]
O último sítio e talvez o mais peculiar da glicogênio fosforilase é o chamado "sítio de armazenamento de glicogênio". Esta estrutura é formada pelos resíduos 397-437, e permite que a proteína se ligue covalentemente à cadeia de glicogênio a cerca de 30 Å do sítio catalítico. Este sítio é muito provavelmente o sítio no qual o enzima se liga aos grânulos de glicogênio antes de iniciar a clivagem das glicoses terminais. De facto, 70% da fosforilase dimérica da célula está ligada aos grânulos de glicogênio em vez de a flutuar livre no citoplasma.[7]
Nos mamíferos, os principais isoenzimas da glicogênio fosforilase encontram-se no músculo, fígado e cérebro. O tipo cerebral é o que predomina no cérebro adulto e nos tecidos embrionários, e os tipos hepático e muscular são os predominantes no fígado adulto e músculo esquelético, respetivamente.[8]
Importância clínica
Propôs-se utilizar a inibição da glicogênio fosforilase como um método para o tratamento da diabetes tipo 2.[9] Como a produção de glicose no fígado viu-se que aumenta nos pacientes com diabetes tipo 2,[10] a inibição da libertação de glicose da reserva de glicogênio hepática pode ser um bom tratamento. A clonagem da glicogênio fosforilase hepática humana (HLGP) revelou um novo sítio de ligação alostérica perto da interface da subunidade, que não está presente na glicogênio fosforilase muscular de coelho (RMGP) utilizada normalmente nos estudos. Este sítio não era sensível aos mesmos inibidores que o sítio alostérico para o AMP,[11] e o maior sucesso na inibição conseguiu-se sintetizando novos inibidores que imitam a estrutura da glicose, já que a glicose 6-fosfato é um inibidor conhecido da HLPG e estabiliza o estado T, que é menos ativo.[12] Estes derivados da glicose tiveram algum sucesso inibindo a HLPG, com uns baixos valores previstos de Ki de 0,016 mM.[13]
As mutações na isoforma do músculo (PYGM) estão associadas com a doença de McArdle (doença de armazenamento de glicogênio tipo V). Identificaram-se até agora mais de 65 mutações no gene da PYGM que originam a doença de McArdle.[14][15] Entre os sintomas da doença de McArdle estão a fraqueza muscular, mialgia, e falta de resistência, todos devidos aos baixos níveis de glicose no tecido muscular.[16]
As mutações na isoforma hepática da glicogênio fosforilase (PYGL) estão associadas com a doença de Hers (doença de armazenamento de glicogênio de tipo VI).[17][18] A doença de Hers está frequentemente associada com sintomas leves normalmente limitados a hipoglicemia, e às vezes é difícil de diagnosticar devido à atividade enzimática residual.[19]
A isoforma cerebral da glicogênio fosforilase (PYGLB) propôs-se como biomarcador do cancro de estômago.[20]
Regulação Alostérica
Músculo
Quando o músculo está em repouso ele está na sua configuração inativa, ou seja, na forma de fosforilase b, mas quando há uma grande atividade muscular a fosforilase b é transformada pela adrenalina em fosforilase ɑ, isto é, na forma mais ativa.
A transformação de fosforilase ɑ em fosforilase b é feita por perda enzimática dos resíduos Ser14 , reação feita pela fosforilase-ɑ-fosfatase. A reação inversa é feita pela fosforilase-b-cinase. O cálcio também é um indutor da fosforilase-b-cinase, fazendo com que o glicogênio fosforilase seja mais ativo (figura 4).[21]
Fígado
Assim como no músculo, a fosforilase ɑ é mais ativa e mais fosforilada que a b devido a baixa glicose. Esse pequeno nível de glicose no sangue faz com que o glucagon ative a fosforilase-b-cinase que converte a fosforilase b em fosforilase ɑ para que então a glicose vá para o sangue. A glicose quando não está mais em baixa quantidade, seu nível considerado normal, entra nos hepatócitos para poder se ligar a um sítio inibitório na fosforilase ɑ expondo os resíduos fosforilados de Ser para PP1(fosfoproteína-fosfatase 1) (Figura 5). [21]
Regulação hormonal
A glicogênio fosforilase está regulada por controlo alostérico e por fosforilação.
As hormonas como a adrenalina (epinefrina), a insulina e o glucagon regulam a glicogênio fosforilase utilizando sistemas de amplificação de segundo mensageiro que estão associados a proteínas G. A adrenalina ativa a adenilato ciclase por meio de um receptor acoplado à proteína G, que por sua vez ativa a adenilato ciclase fazendo aumentar as concentrações celulares de AMP cíclico (AMPc). O AMPc liga-se (e depois liberta-se) a uma forma ativa de proteína quinase A (PKA). Depois, a proteína quinase A fosforila a fosforilase quinase, que por sua vez fosforila a glicogênio fosforilase b, transformando-a na glicogênio fosforilase a ativa. Esta fosforilação realiza-se sobre a serina 14 da glicogênio fosforilase b. No fígado, o glucagon ativa outro receptor acoplado à proteína G que desencadeia outra cascata de reações diferente, causando a ativação da fosfolipase C (PLC). A fosfolipase C causa indiretamente a libertação de cálcio do retículo endoplasmático dos hepatócitos no citosol. O aumento da disponibilidade de cálcio faz com que este se ligue à subunidade calmodulina e ative a glicogênio fosforilase quinase. A glicogênio fosforilase quinase ativa a glicogênio fosforilase da mesma maneira mencionada previamente.
A glicogênio fosforilase b nem sempre é inativa no músculo, já que pode ser ativada alostericamente pelo AMP. Um aumento na concentração de AMP, como a que tem lugar durante o exercício extenuante, indica que há uma grande procura de energia. O AMP ativa a glicogênio fosforilase b mudando a sua conformação de uma forma tensa (T) para uma forma relaxada (R). Esta forma relaxada tem propriedades enzimáticas similares às do enzima fosforilado. Um aumento na concentração de ATP opõe-se a esta ativação ao deslocar o AMP do sítio de ligação para o nucleótido no enzima, indicando que há reservas de energia suficientes.
Depois de comer há uma libertação de insulina, o que indica que há uma grande quantidade de glicose no sangue. A insulina ativa indiretamente a PP-1 e a fosfodiesterase. A proteína fosfatase 1 (PP-1) desfosforila diretamente a glicogênio fosforilase a, voltando a originar-se a glicogênio fosforilase b inativa. A fosfodiesterase converte o AMPc em AMP. Esta atividade elimina o segundo mensageiro (gerado pelo glucagon e pela adrenalina) e inibe a proteína quinase A. Desta forma, a proteína quinase A já não pode causar a cascata de fosforilação que acaba com a formação de glicogênio fosforilase a (ativa). Estas modificações iniciadas pela insulina detêm a glicogenólise para preservar as reservas de glicogênio que ficam na célula e desencadear a glicogenogénese (formação de glicogênio).
As fosforilases a e b podem existir em duas formas, uma delas o estado T (tenso) inativo e a outra o estado R (relaxado). A fosforilase b está normalmente no estado T, inativa devido à presença fisiológica de ATP e glicose 6-fosfato, e a fosforilase a está normalmente no estado R (ativa).
No fígado existe um isoenzima da glicogênio fosforilase sensível às concentrações de glicose, já que o fígado atua como exportador de glicose. Em essência, a fosforilase do fígado é sensível à glicose, o que causa uma transição muito sensível da forma R à T, inativando-a; além disso, a fosforilase hepática é insensível ao AMP.
Referências
- ↑ a b Livanova NB, Chebotareva NA, Eronina TB, Kurganov BI (2002). «Pyridoxal 5′_Phosphate as a Catalytic and Conformational Cofactor of Muscle Glycogen Phosphorylase b». Biochemistry (Moscow). 67 (10): 1089–1998. PMID 12460107. doi:10.1023/A:1020978825802
- ↑ Palm D, Klein HW, Schinzel R, Buehner M, Helmreich, EJM (1990). «The role of pyridoxal 5'-phosphate in glycogen phosphorylase catalysis». Biochemistry. 29 (5): 1099–1107. PMID 2182117. doi:10.1021/bi00457a001
- ↑ Browner MF, Fletterick RJ (1992). «Phosphorylase: a biological transducer». Trends in Biochemical Science. 17 (2): 66–71. PMID 1566331. doi:10.1016/0968-0004(92)90504-3
- ↑ a b Johnson LN (1992). «Glycogen phosphorylase: control by phosphorylation and allosteric effectors». FASEB Journal. 6 (6): 2274–82. PMID 1544539
- ↑ Newgard CB, Hwang PK, Fletterick, RJ (1989). «The family of glycogen phosphorylases: structure and function». Critical Reviews Biochemistry and Molecular Biology. 24 (1): 69–99. PMID 2667896. doi:10.3109/10409238909082552
- ↑ Johnson LN, Barford, D (1990). «Glycogen phosphorylase. The structural basis of the allosteric response and comparison with other allosteric proteins.». Journal of Biological Chemistry. 265 (5): 2409–2412. PMID 2137445
- ↑ Meyer F, Heilmeyer LM Jr, Haschke RH, Fischer EH (1970). «Control of phosphorylase activity in a muscle glycogen particle. I. Isolation and characterization of the protein-glycogen complex». Journal of Biological Chemistry. 245 (24): 6642–6648. PMID 4320610
- ↑ David ES, Crerar MM (1986). «Quantitation of muscle glycogen phosphorylase mRNA and enzyme amounts in adult rat tissues». Biochim. Biophys. Acta. 880 (1): 78–90. PMID 3510670
- ↑ Somsák L, Nagya V, Hadady Z, Docsa T, Gergely P. (2003). «Glucose analog inhibitors of glycogen phosphorylases as potential antidiabetic agents: recent developments». Current Pharmacological Design. 9 (15): 1177–89. PMID 12769745. doi:10.2174/1381612033454919
- ↑ Moller DE (2001). «New drug targets for type 2 diabetes and the metabolic syndrome». Nature. 414 (6865): 821–7. PMID 11742415. doi:10.1038/414821a
- ↑ Coats WS, Browner MF, Fletterick RJ, Newgard CB (1991). «An engineered liver glycogen phosphorylase with AMP allosteric activation». Journal of Biological Chemistry. 266 (24): 16113–9. PMID 1874749
- ↑ Oikonomakos NG, Kontou M, Zographos SE, Tsitoura HS, Johnson LN, Watson KA, Mitchell EP, Fleet GW, Son JC, Bichard CJ; et al. (1994). «The design of potential antidiabetic drugs: experimental investigation of a number of beta-D-glucose analogue inhibitors of glycogen phosphorylase». European Journal of Drug Metabolism and Pharmacology. 19 (3): 185–92. PMID 7867660. doi:10.1007/BF03188920
- ↑ Hopfinger A J, Reaka A, Venkatarangan P, Duca J S, Wang S. (1999). «Prediction of Ligand−Receptor Binding Free Energy by 4D-QSAR Analysis: Application to a Set of Glucose Analogue Inhibitors of Glycogen Phosphorylase». Journal of Chemical Information and Computer Science. 39: 1141–1150. doi:10.1021/ci9900332
- ↑ Nogales-Gadea G, Arenas J, Andreu AL (2007). «Molecular genetics of McArdle's disease». Curr Neurol Neurosci Rep. 7 (1): 84–92. PMID 17217859. doi:10.1007/s11910-007-0026-2
- ↑ Andreu AL, Nogales-Gadea G, Cassandrini D, Arenas J, Bruno C (2007). «McArdle disease: molecular genetic update». Acta Myol. 26 (1): 53–7. PMID 17915571
- ↑ Grünfeld JP, Ganeval D, Chanard J, Fardeau M, Dreyfus JC (1972). «Acute renal failure in McArdle's disease. Report of two cases». New England Journal of Medicine. 286 (23): 1237–41. PMID 4502558. doi:10.1056/NEJM197206082862304
- ↑ Burwinkel B, Bakker HD, Herschkovitz E, Moses SW, Shin YS, Kilimann MW (1998). «Mutations in the liver glycogen phosphorylase gene (PYGL) underlying glycogenosis type VI». Am. J. Hum. Genet. 62 (4): 785–91. PMC 1377030
 . PMID 9529348. doi:10.1086/301790
. PMID 9529348. doi:10.1086/301790
- ↑ Chang S, Rosenberg MJ, Morton H, Francomano CA, Biesecker LG (1998). «Identification of a mutation in liver glycogen phosphorylase in glycogen storage disease type VI». Hum. Mol. Genet. 7 (5): 865–70. PMID 9536091. doi:10.1093/hmg/7.5.865
- ↑ Tang NL, Hui J, Young E, Worthington V, To KF, Cheung KL, Li CK, Fok TF (2003). «A novel mutation (G233D) in the glycogen phosphorylase gene in a patient with hepatic glycogen storage disease and residual enzyme activity». Molecular Genetics and Metabolism. 79 (2): 142–145. PMID 12809646. doi:10.1016/S1096-7192(03)00068-4
- ↑ Shimada S, Matsuzaki H, Marutsuka T, Shiomori K, Ogawa M (2001). «Gastric and intestinal phenotypes of gastric carcinoma with reference to expression of brain (fetal)-type glycogen phosphorylase». J. Gastroenterol. 36 (7): 457–64. PMID 11480789. doi:10.1007/s005350170068
- ↑ a b Erro de citação: Etiqueta
<ref>inválida; não foi fornecido texto para as "refs" nomeadas:0