Terapia com anticorpos monoclonais
Os anticorpos monoclonais (mAbs) têm usos terapêuticos variados. É possível criar um mAb que se liga especificamente a quase qualquer alvo extracelular, como proteínas de superfície celular e citocinas . Eles podem ser usados para tornar seu alvo ineficaz (por exemplo, impedindo a ligação ao receptor), [1] para induzir um sinal celular específico (ativando receptores), [1] para fazer com que o sistema imunológico ataque células específicas ou para levar um medicamento a um tipo específico de célula (como com a radioimunoterapia que administra radiação citotóxica ).
As principais aplicações incluem câncer, doenças autoimunes, asma, transplantes de órgãos, prevenção de coágulos sanguíneos e certas infecções.
Estrutura e função do anticorpo
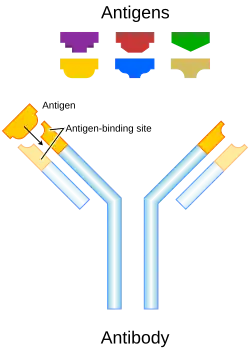
Anticorpos da imunoglobulina G (IgG) são grandes moléculas heterodiméricas, aproximadamente 150 kDa e são compostos de dois tipos de cadeia polipeptídica, chamadas de pesadas (~50 kDa) e cadeias leves (~25 kDa). Os dois tipos de cadeias leves são kappa (κ) e lambda (λ). Por clivagem com a enzima papaína, a parte Fab (ligação fragmento-antígeno) pode ser separada da parte Fc (região cristalizável do fragmento) da molécula. Os fragmentos Fab contêm os domínios variáveis, que consistem em três domínios de aminoácidos hipervariáveis do anticorpo responsáveis pela especificidade do anticorpo embutidos em regiões constantes. As quatro subclasses conhecidas de IgG estão envolvidas na citotoxicidade celular dependente de anticorpos.[2] Os anticorpos são um componente chave da resposta imune adaptativa, desempenhando um papel central tanto no reconhecimento de antígenos estranhos quanto na estimulação de uma resposta imune a eles. O advento da tecnologia de anticorpos monoclonais tornou possível criar anticorpos contra antígenos específicos presentes nas superfícies dos tumores.[3] Os anticorpos monoclonais podem ser adquiridos no sistema imunológico por meio da imunidade passiva ou imunidade ativa. A vantagem da terapia com anticorpos monoclonais ativos é o fato de que o sistema imunológico produzirá anticorpos a longo prazo, com apenas uma administração de medicamentos de curto prazo para induzir essa resposta. No entanto, a resposta imunológica a certos antígenos pode ser inadequada, especialmente em idosos. Além disso, reações adversas desses anticorpos podem ocorrer devido a uma resposta prolongada a antígenos.[4] A terapia com anticorpos monoclonais passivos pode garantir uma concentração consistente de anticorpos e pode controlar as reações adversas por meio da interrupção da administração. No entanto, a administração repetida e o consequente custo mais alto dessa terapia são as principais desvantagens. [4]
A terapia com anticorpos monoclonais pode vir a ser benéfica para câncer, doenças autoimunes e distúrbios neurológicos que resultam na degeneração de células do corpo, como a doença de Alzheimer . A terapia com anticorpos monoclonais pode ajudar o sistema imunológico porque o sistema imunológico inato responde aos fatores ambientais que encontra diferenciando células estranhas das células do corpo. Portanto, células tumorais que estão proliferando em altas taxas, ou células do corpo que estão morrendo, o que subsequentemente causa problemas fisiológicos, geralmente não são alvos específicos do sistema imunológico, uma vez que as células tumorais são células do próprio paciente. As células tumorais, no entanto, são altamente anormais e muitas exibem antígenos incomuns. Alguns desses antígenos tumorais são inapropriados para o tipo de célula ou seu ambiente. Os anticorpos monoclonais podem ter como alvo células tumorais ou células anormais no corpo que são reconhecidas como células do corpo, mas são debilitantes para a saúde.[carece de fontes]
Histórico
A imunoterapia foi desenvolvida na década de 1970 após a descoberta da estrutura dos anticorpos e o desenvolvimento da tecnologia do hibridoma, que forneceu a primeira fonte confiável de anticorpos monoclonais.[6][7] Esses avanços permitiram o direcionamento específico de tumores in vitro e in vivo. A pesquisa inicial sobre neoplasias malignas encontrou na terapia com mAb um sucesso limitado e geralmente de curta duração em malignidades sanguíneas.[8][9] O tratamento também teve que ser adaptado a cada paciente, o que era impraticável em ambientes clínicos de rotina.[carece de fontes]
Os quatro principais tipos de anticorpos desenvolvidos são murinos, quiméricos, humanizados e humanos. Os anticorpos de cada tipo são distinguidos por sufixos em seus nomes.[carece de fontes]
Murinos
Os anticorpos terapêuticos iniciais eram análogos murinos (sufixo -omab ). Esses anticorpos têm: meia-vida curta in vivo (devido à formação de complexos imunes), penetração limitada em locais tumorais e recrutam inadequadamente funções efetoras do hospedeiro.[10] Anticorpos quiméricos e humanizados os substituíram em geral para aplicações de anticorpos terapêuticos.[11] A compreensão da proteômica provou ser essencial na identificação de novos alvos tumorais.[carece de fontes]
Inicialmente, os anticorpos murinos foram obtidos pela tecnologia de hibridoma, pela qual Jerne, Köhler e Milstein receberam um prêmio Nobel. No entanto, a diferença entre os sistemas imunológicos murino e humano levou à falha clínica desses anticorpos, exceto em algumas circunstâncias específicas. Os principais problemas associados aos anticorpos murinos incluíram a redução da estimulação da citotoxicidade e a formação de complexos após administração repetida, o que resultou em reações alérgicas leves e, às vezes, choque anafilático.[10] A tecnologia de hibridoma foi substituída pela tecnologia de DNA recombinante, camundongos transgênicos e display de fagos.[11]
Quiméricos e humanizados
Para reduzir a imunogenicidade do anticorpo murino (ataques do sistema imunológico contra o anticorpo), moléculas murinas foram projetadas para remover o conteúdo imunogênico e aumentar a eficiência imunológica. [10] Isso foi alcançado inicialmente pela produção de anticorpos quiméricos (sufixo -ximab) e humanizados (sufixo -zumab ). Os anticorpos quiméricos são compostos de regiões variáveis murinas fundidas em regiões constantes humanas. A retirada de sequências de genes humanos da cadeia leve kappa e da cadeia pesada IgG1 resulta em anticorpos que são aproximadamente 65% humanos. Isso reduz a imunogenicidade e, portanto, aumenta a meia-vida sérica.[carece de fontes]
Os anticorpos humanizados são produzidos por enxerto de regiões hipervariáveis murinas em domínios de aminoácidos em anticorpos humanos. Isso resulta em uma molécula de aproximadamente 95% de origem humana. Os anticorpos humanizados ligam-se ao antígeno de forma muito mais fraca do que o anticorpo monoclonal murino original, com reduções relatadas na afinidade de até várias centenas de vezes. [12][13] Aumentos na força de ligação anticorpo-antígeno foram alcançados pela introdução de mutações nas regiões determinantes de complementariedade (CDR), [14] usando técnicas como embaralhamento de cadeia, randomização de regiões determinantes de complementariedade e anticorpos com mutações dentro das regiões variáveis induzidas por ep-PCR (PCR propensa a erros), cepas mutadoras de E. coli e mutagênese específica de sítio . [15]
Anticorpos monoclonais humanos
Os anticorpos monoclonais humanos (sufixo -umab ) são produzidos usando camundongos transgênicos ou bibliotecas display de fagos, transferindo genes de imunoglobulina humana para o genoma murino e vacinando o camundongo transgênico contra o antígeno desejado, levando à produção de anticorpos monoclonais apropriados.[11] Os anticorpos murinos in vitro são, portanto, transformados em anticorpos totalmente humanos.[3]
As cadeias pesadas e leves das proteínas IgG humanas são expressas em formas polimórficas estruturais (alotípicas). O alótipo da IgG humana é um dos muitos fatores que podem contribuir para a imunogenicidade.[16][17]
Condições alvo
Câncer
Os anticorpos monoclonais anticâncer podem ser direcionados contra células malignas por vários mecanismos. O ramucirumabe é um anticorpo monoclonal humano recombinante e é usado no tratamento de malignidades avançadas. [18] No linfoma infantil, estudos de fase I e II encontraram um efeito positivo do uso da terapia com anticorpos. [19]
Anticorpos monoclonais usados para impulsionar uma resposta imune anticâncer são outra estratégia para combater o câncer onde as células cancerígenas não são alvos diretos. As estratégias incluem anticorpos projetados para bloquear mecanismos que regulam negativamente as respostas imunes anticâncer, pontos de verificação como PD-1 e CTLA-4 (terapia de checkpoint), [20] e anticorpos modificados para estimular a ativação de células imunes. [21]
Doenças autoimunes
Os anticorpos monoclonais usados para doenças autoimunes incluem infliximab e adalimumab, que são eficazes na artrite reumatoide, doença de Crohn e colite ulcerativa por sua capacidade de se ligar e inibir o TNF-α . [22] Basiliximab e daclizumab inibem a IL-2 em células T ativadas e, assim, ajudam a prevenir a rejeição aguda de transplantes renais.[22] O omalizumab inibe a imunoglobulina E humana (IgE) e é útil na asma alérgica moderada à grave.[carece de fontes]
Doença de Alzheimer
A doença de Alzheimer (DA) é uma doença neurodegenerativa progressiva, multifacetada e dependente da idade, e é uma das principais causas de demência. [23] De acordo com a hipótese amilóide, o acúmulo de peptídeos beta amilóides extracelulares (Aβ) em placas por meio de oligomerização leva a condições sintomáticas características da DA por meio da disfunção sináptica e neurodegeneração. [24] A imunoterapia por meio da administração de anticorpo monoclonal exógeno (mAb) é conhecida por tratar vários distúrbios do sistema nervoso central. No caso da DA, acredita-se que a imunoterapia iniba a oligomerização de Aβ ou remova Aβ do cérebro e, assim, previna a neurotoxicidade . [25]
No entanto, os mAbs são moléculas grandes e, devido à barreira hematoencefálica, a absorção de mAbs no cérebro é extremamente limitada, estima-se que apenas aproximadamente 1 em cada 1000 moléculas de mAbs passe. [25] No entanto, a hipótese do "sink periférico" propõe um mecanismo em que os mAbs podem não precisar atravessar a barreira hematoencefálica. [26] Portanto, muitos estudos de pesquisa estão sendo conduzidos a partir de tentativas fracassadas de tratar a DA no passado. [24]
No entanto, as vacinas anti-Aβ podem promover a eliminação mediada por anticorpos das placas Aβ em modelos de camundongos transgênicos com proteínas precursoras de amiloide (APP) e podem reduzir deficiências cognitivas. [23] As vacinas podem estimular o sistema imunológico a produzir seus próprios anticorpos, no caso da doença de Alzheimer pela administração do antígeno Aβ. [27] Isso também é conhecido como imunoterapia ativa . Outra estratégia é a chamada imunoterapia passiva . Nesse caso, os anticorpos são produzidos externamente em células cultivadas e são entregues ao paciente na forma de um medicamento. Em camundongos que expressam APP, a imunização ativa e passiva de anticorpos anti-Aβ demonstrou ser eficaz na eliminação de placas e pode melhorar a função cognitiva. [24]
Atualmente, existem duas terapias de anticorpos aprovadas pela FDA para a doença de Alzheimer, Aducanemab e Lecanemab . O Aducanemab recebeu aprovação acelerada, enquanto o Lecanemab recebeu aprovação total. [25] Vários ensaios clínicos usando imunização passiva e ativa foram realizados e alguns estão em andamento com resultados esperados em alguns anos. [24] [25] A implementação dessas drogas geralmente ocorre durante o início precoce da DA. Um ensaio que testa o Foralumab busca determinar se há benefício em estágios posteriores da DA pela redução da inflamação cerebral. [28] Outras pesquisas e desenvolvimento de drogas para intervenção precoce e prevenção da DA estão em andamento. Exemplos de drogas mAb importantes que foram ou estão sob avaliação para o tratamento da DA incluem Bapineuzumab, Solanezumab, Gautenerumab, Crenezumab, Aducanemab, Lecanemab e Donanemab . [25]
Bapineuzumab
Bapineuzumab, um mAb anti-Aβ humanizado, é direcionado contra o N-terminal de Aβ. Os ensaios clínicos de fase II de Bapineuzumab em pacientes com DA leve à moderada resultaram em concentração reduzida de Aβ no cérebro. No entanto, em pacientes com carreadores aumentados de apolipoproteína (APOE) e4, o tratamento com Bapineuzumab também é acompanhado por edema vasogênico, [29] uma condição citotóxica em que a barreira hematoencefálica foi rompida, afetando assim a substância branca devido ao acúmulo excessivo de fluido dos capilares nos espaços intracelulares e extracelulares do cérebro. [30]
Em ensaios clínicos de Fase III, o Bapineuzumabe demonstrou um efeito positivo promissor em biomarcadores da DA, mas não demonstrou efeito no declínio cognitivo. Portanto, o Bapineuzumabe foi descontinuado após falha no ensaio clínico de Fase III. [30]
Solanezumab
Solanezumab, um mAb anti-Aβ, tem como alvo o N-terminal de Aβ. Na Fase I e Fase II dos ensaios clínicos, o tratamento com Solanezumab resultou na elevação do líquido cefalorraquidiano de Aβ, mostrando assim uma concentração reduzida de placas de Aβ. Além disso, não há efeitos colaterais adversos associados. Os ensaios clínicos de Fase III de Solanezumab trouxeram uma redução significativa no comprometimento cognitivo em pacientes com DA leve, mas não em pacientes com DA grave. No entanto, a concentração de Aβ não mudou significativamente, juntamente com outros biomarcadores de DA, incluindo expressão de fosfo-tau e volume hipocampal. Os ensaios clínicos de Fase III de Solanezumab falharam, pois não mostraram efeito no declínio cognitivo em comparação ao placebo.[31]
Lecanemab
Lecanemab (BAN2401), é um mAb humanizado que tem como alvo seletivo protofibrilas Aβ solúveis tóxicas, [32] Em ensaios clínicos de fase 3, [33] o Lecanemab mostrou um declínio cognitivo 27% mais lento após 18 meses de tratamento em comparação com placebo. [34] [35] Os ensaios clínicos de fase 3 também relataram reações relacionadas à infusão, anormalidades de imagem relacionadas ao amiloide e dores de cabeça foram os efeitos colaterais mais comuns do Lecanemab. Em julho de 2023, o FDA deu ao Lecanemab aprovação total para o tratamento da doença de Alzheimer [36] e ele recebeu o nome comercial de Leqembi.
Ensaios preventivos
A falha de vários medicamentos em ensaios clínicos de Fase III levou à prevenção da DA e à intervenção precoce para esforços de tratamento da DA inicial. O tratamento passivo com mAb anti-Aβ pode ser usado para tentativas preventivas de modificar a progressão da DA antes que ela cause danos cerebrais e sintomas extensos. Ensaios usando tratamento com mAb para pacientes positivos para fatores de risco genéticos e pacientes idosos positivos para indicadores de DA estão em andamento. Isso inclui o tratamento anti-AB na Doença de Alzheimer Assintomática (A4), a Iniciativa de Prevenção de Alzheimer (API) e DIAN-TU. [26] O estudo A4 em indivíduos mais velhos que são positivos para indicadores de DA, mas são negativos para fatores de risco genéticos testará o Solanezumab em Ensaios Clínicos de Fase III, como um acompanhamento de estudos anteriores com Solanezumab. [26] O DIAN-TU, lançado em dezembro de 2012, foca em pacientes jovens positivos para mutações genéticas que são riscos para DA. Este estudo usa Solanezumab e Gautenerumab. Gautenerumab, o primeiro MAB totalmente humano que interage preferencialmente com placas de Aβ oligomerizadas no cérebro, causou redução significativa na concentração de Aβ em ensaios clínicos de Fase I, prevenindo a formação e a concentração de placas sem alterar a concentração plasmática no cérebro. Ensaios clínicos de Fase II e III estão sendo conduzidos atualmente. [26]
Tipos de terapia
Radioimunoterapia
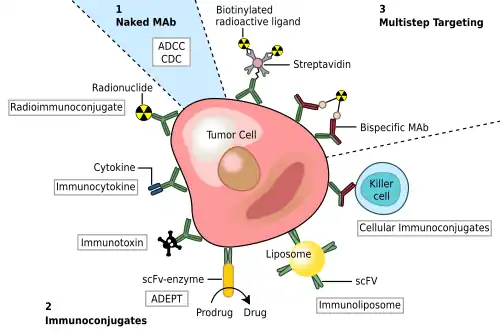
A radioimunoterapia (RIT) envolve o uso de anticorpos murinos conjugados radioativamente contra antígenos celulares. A maioria das pesquisas envolve sua aplicação em linfomas, visto que estes são tumores malignos altamente radiossensíveis. Para limitar a exposição à radiação, os anticorpos murinos foram escolhidos, pois sua alta imunogenicidade promove a rápida eliminação do tumor. O tositumomab é um exemplo usado para o linfoma não Hodgkin.
Terapia enzimática com pró-fármaco dirigida por anticorpo
A terapia enzimática com pró-fármacos dirigida por anticorpos (ADEPT) envolve a aplicação de anticorpos monoclonais associados ao câncer, que estão ligados a uma enzima ativadora de fármacos. A administração sistêmica de um agente não tóxico resulta na conversão do anticorpo em um fármaco tóxico, resultando em um efeito citotóxico que pode ser direcionado às células malignas. O sucesso clínico dos tratamentos com ADEPT é limitado. [37]
Conjugados anticorpo-fármaco
Conjugados anticorpo-fármaco (ADCs) são anticorpos ligados a uma ou mais moléculas de fármaco. Normalmente, quando o ADC encontra a célula-alvo (por exemplo, uma célula cancerosa), o fármaco é liberado para matá-la. Muitos ADCs estão em desenvolvimento clínico. Desde 2016 alguns foram aprovados.[carece de fontes]
Terapia imunolipossomal
Os imunolipossomas são lipossomas conjugados a anticorpos. Os lipossomas podem transportar fármacos ou nucleotídeos terapêuticos e, quando conjugados com anticorpos monoclonais, podem ser direcionados contra células malignas. Os imunolipossomas têm sido utilizados com sucesso in vivo para transportar genes supressores de tumores para os tumores, utilizando um fragmento de anticorpo contra o receptor de transferrina humana. A administração de genes específicos para tecidos utilizando imunolipossomas foi alcançada em tecidos de câncer cerebral e de mama. [38]
Terapia 'checkpoint'
A terapia checkpoint utiliza anticorpos e outras técnicas para contornar as defesas que os tumores usam para suprimir o sistema imunológico. Cada defesa é conhecida como um checkpoint. Terapias compostas combinam anticorpos para suprimir múltiplas camadas defensivas. Os checkpoints conhecidos incluem CTLA-4, alvo do ipilimumab, PD-1, alvo do nivolumab e pembrolizumabe, e o microambiente tumoral. [20]
As características do microambiente tumoral (TME) impedem o recrutamento de células T para o tumor. As formas incluem a nitração da quimiocina CCL 2, que aprisiona as células T no estroma . A vasculatura tumoral ajuda os tumores a recrutar preferencialmente outras células imunes em vez das células T, em parte por meio da expressão específica de células endoteliais (EC) de FasL, ET B R e B7H3. Células mielomonocíticas e tumorais podem regular positivamente a expressão de PD-L1, em parte impulsionada por condições hipóxicas e pela produção de citocinas, como IFNβ. A produção aberrante de metabólitos no TME, como a regulação da via por IDO, pode afetar as funções das células T direta e indiretamente por meio de células como T reg. células. As células CD8 podem ser suprimidas pela regulação dos fenótipos TAM pelas células B. Os fibroblastos associados ao câncer (CAFs) têm múltiplas funções de TME, em parte por meio do aprisionamento de células T mediado pela matriz extracelular (ECM) e da exclusão de células T reguladas por CXCL12 . [39]
Anticorpos terapêuticos aprovados pela FDA
O primeiro anticorpo monoclonal terapêutico aprovado pela FDA foi um medicamento murino IgG2a CD3 específico para rejeição de transplantes, o OKT3 (também chamado de muromonab), em 1986. Este medicamento foi utilizado em beneficiários de transplantes de órgãos sólidos que se tornaram resistentes a esteroides . [40] Centenas de terapias estão em fase de ensaios clínicos . A maioria se concentra em alvos imunológicos e oncológicos.
Economia
Desde 2000, o mercado terapêutico para anticorpos monoclonais cresceu exponencialmente. Em 2006, os "5 grandes" anticorpos terapêuticos no mercado eram bevacizumab, trastuzumabe (ambos oncológicos), adalimumabe, infliximabe (ambos para doenças autoimunes e inflamatórias, 'AIID') e rituximab (oncologia e AIID) que representaram 80% das receitas em 2006. Em 2007, oito dos 20 medicamentos de biotecnologia mais vendidos nos EUA eram anticorpos monoclonais terapêuticos. [41] Esse rápido crescimento na demanda pela produção de anticorpos monoclonais foi bem acomodado pela industrialização da fabricação de mAb. [42]
Referências
- ↑ a b Yao S, Zhu Y, Chen L (fevereiro de 2013). «Advances in targeting cell surface signalling molecules for immune modulation». Nature Reviews. Drug Discovery. 12 (2): 130–146. PMC 3698571
 . PMID 23370250. doi:10.1038/nrd3877
. PMID 23370250. doi:10.1038/nrd3877
- ↑ Janeway, Charles; Paul Travers; Mark Walport; Mark Shlomchik (2001). Immunobiology; Fifth Edition. New York and London: Garland Science. ISBN 978-0-8153-4101-7
- ↑ a b Janeway CA Jr.; et al. (2005). Immunobiology 6th ed. [S.l.]: Garland Science. ISBN 978-0-443-07310-6
- ↑ a b Baxter D (dezembro de 2007). «Active and passive immunity, vaccine types, excipients and licensing». Occupational Medicine. 57 (8): 552–556. PMID 18045976. doi:10.1093/occmed/kqm110
- ↑ Modified from Carter P (novembro de 2001). «Improving the efficacy of antibody-based cancer therapies». Nature Reviews. Cancer. 1 (2): 118–129. PMID 11905803. doi:10.1038/35101072
- ↑ Breedveld FC (fevereiro de 2000). «Therapeutic monoclonal antibodies». Lancet. 355 (9205): 735–740. PMID 10703815. doi:10.1016/S0140-6736(00)01034-5
- ↑ Köhler G, Milstein C (agosto de 1975). «Continuous cultures of fused cells secreting antibody of predefined specificity». Nature. 256 (5517): 495–497. Bibcode:1975Natur.256..495K. PMID 1172191. doi:10.1038/256495a0
- ↑ Nadler LM, Stashenko P, Hardy R, Kaplan WD, Button LN, Kufe DW, et al. (setembro de 1980). «Serotherapy of a patient with a monoclonal antibody directed against a human lymphoma-associated antigen». Cancer Research. 40 (9): 3147–3154. PMID 7427932
- ↑ Ritz J, Schlossman SF (janeiro de 1982). «Utilization of monoclonal antibodies in the treatment of leukemia and lymphoma». Blood. 59 (1): 1–11. PMID 7032624. doi:10.1182/blood.V59.1.1.1
- ↑ a b c Stern M, Herrmann R (abril de 2005). «Overview of monoclonal antibodies in cancer therapy: present and promise». Critical Reviews in Oncology/Hematology. 54 (1): 11–29. PMID 15780905. doi:10.1016/j.critrevonc.2004.10.011
- ↑ a b c Hudson PJ, Souriau C (janeiro de 2003). «Engineered antibodies». Nature Medicine. 9 (1): 129–134. PMID 12514726. doi:10.1038/nm0103-129
- ↑ Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, et al. (maio de 1992). «Humanization of an anti-p185HER2 antibody for human cancer therapy». Proceedings of the National Academy of Sciences of the United States of America. 89 (10): 4285–4289. Bibcode:1992PNAS...89.4285C. PMC 49066. PMID 1350088. doi:10.1073/pnas.89.10.4285
- ↑ Presta LG, Lahr SJ, Shields RL, Porter JP, Gorman CM, Fendly BM, Jardieu PM (setembro de 1993). «Humanization of an antibody directed against IgE». Journal of Immunology. 151 (5): 2623–2632. PMID 8360482. doi:10.4049/jimmunol.151.5.2623
- ↑ Chothia C, Lesk AM, Tramontano A, Levitt M, Smith-Gill SJ, Air G, et al. (1989). «Conformations of immunoglobulin hypervariable regions». Nature. 342 (6252): 877–883. Bibcode:1989Natur.342..877C. PMID 2687698. doi:10.1038/342877a0
- ↑ Waldmann TA (março de 2003). «Immunotherapy: past, present and future». Nature Medicine. 9 (3): 269–277. PMID 12612576. doi:10.1038/nm0303-269
- ↑ Jefferis R, Lefranc MP (julho–agosto de 2009). «Human immunoglobulin allotypes: possible implications for immunogenicity». mAbs. 1 (4): 332–338. PMC 2726606. PMID 20073133. doi:10.4161/mabs.1.4.9122
- ↑ Chapman K, Pullen N, Coney L, Dempster M, Andrews L, Bajramovic J, et al. (2009). «Preclinical development of monoclonal antibodies: considerations for the use of non-human primates». mAbs. 1 (5): 505–516. PMC 2759500. PMID 20065651. doi:10.4161/mabs.1.5.9676
- ↑ Vennepureddy A, Singh P, Rastogi R, Atallah JP, Terjanian T (outubro de 2017). «Evolution of ramucirumab in the treatment of cancer - A review of literature». Journal of Oncology Pharmacy Practice. 23 (7): 525–539. PMID 27306885. doi:10.1177/1078155216655474
- ↑ de Zwart V, Gouw SC, Meyer-Wentrup FA (janeiro de 2016). «Antibody therapies for lymphoma in children». The Cochrane Database of Systematic Reviews. 2016 (1): CD011181. PMC 8719646. PMID 26784573. doi:10.1002/14651858.cd011181.pub2
- ↑ a b Sharma P, Allison JP (abril de 2015). «The future of immune checkpoint therapy». Science. 348 (6230): 56–61. Bibcode:2015Sci...348...56S. PMID 25838373. doi:10.1126/science.aaa8172
- ↑ Dempke WC, Fenchel K, Uciechowski P, Dale SP (março de 2017). «Second- and third-generation drugs for immuno-oncology treatment-The more the better?». European Journal of Cancer. 74: 55–72. PMID 28335888. doi:10.1016/j.ejca.2017.01.001
- ↑ a b Rang, H. P. (2003). Pharmacology. Edinburgh: Churchill Livingstone. ISBN 978-0-443-07145-4
- ↑ a b Pul R, Dodel R, Stangel M (março de 2011). «Antibody-based therapy in Alzheimer's disease». Expert Opinion on Biological Therapy. 11 (3): 343–357. PMID 21261567. doi:10.1517/14712598.2011.552884
- ↑ a b c d van Dyck CH (fevereiro de 2018). «Anti-Amyloid-β Monoclonal Antibodies for Alzheimer's Disease: Pitfalls and Promise». Biological Psychiatry. 83 (4): 311–319. PMC 5767539. PMID 28967385. doi:10.1016/j.biopsych.2017.08.010
- ↑ a b c d e Guo X, Yan L, Zhang D, Zhao Y (fevereiro de 2024). «Passive immunotherapy for Alzheimer's disease». Ageing Research Reviews. 94. 102192 páginas. PMID 38219962. doi:10.1016/j.arr.2024.102192
- ↑ a b c d Panza F, Solfrizzi V, Imbimbo BP, Logroscino G (outubro de 2014). «Amyloid-directed monoclonal antibodies for the treatment of Alzheimer's disease: the point of no return?». Expert Opinion on Biological Therapy. 14 (10): 1465–1476. PMID 24981190. doi:10.1517/14712598.2014.935332
- ↑ van Dyck CH (fevereiro de 2018). «Anti-Amyloid-β Monoclonal Antibodies for Alzheimer's Disease: Pitfalls and Promise». Biological Psychiatry. 83 (4): 311–319. PMC 5767539. PMID 28967385. doi:10.1016/j.biopsych.2017.08.010
- ↑ Hamilton, Jon (30 de maio de 2025). «Can this nasal spray slow down Alzheimer's? One couple is helping scientists find out». NPR (em inglês). Consultado em 30 de maio de 2025
- ↑ Goel, Ayush (20 de agosto de 2013). «Vasogenic cerebral oedema». radiopaedia.org (em inglês). Consultado em 1 de novembro de 2017
- ↑ a b Panza F, Frisardi V, Imbimbo BP, D'Onofrio G, Pietrarossa G, Seripa D, et al. (novembro de 2010). «Bapineuzumab: anti-β-amyloid monoclonal antibodies for the treatment of Alzheimer's disease». Immunotherapy. 2 (6): 767–782. PMID 21091109. doi:10.2217/imt.10.80
- ↑ Sperling RA, Donohue MC, Raman R, Rafii MS, Johnson K, Masters CL, et al. (setembro de 2023). «Trial of Solanezumab in Preclinical Alzheimer's Disease». The New England Journal of Medicine. 389 (12): 1096–1107. PMC 10559996. PMID 37458272. doi:10.1056/NEJMoa2305032
- ↑ Logovinsky V, Satlin A, Lai R, Swanson C, Kaplow J, Osswald G, et al. (abril de 2016). «Safety and tolerability of BAN2401--a clinical study in Alzheimer's disease with a protofibril selective Aβ antibody». Alzheimer's Research & Therapy. 8 (1). 14 páginas. PMC 4822297. PMID 27048170. doi:10.1186/s13195-016-0181-2
- ↑ «A Study to Confirm Safety and Efficacy of BAN2401 in Participants With Early Alzheimer's Disease»
 . Case Medical Research. 25 de março de 2019. ISSN 2643-4652. doi:10.31525/ct1-nct03887455
. Case Medical Research. 25 de março de 2019. ISSN 2643-4652. doi:10.31525/ct1-nct03887455
- ↑ van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. (janeiro de 2023). «Lecanemab in Early Alzheimer's Disease». The New England Journal of Medicine. 388 (1): 9–21. PMID 36449413. doi:10.1056/NEJMoa2212948
- ↑ «Leqembi | ALZFORUM». www.alzforum.org (em inglês). Consultado em 14 de fevereiro de 2024
- ↑ Commissioner, Office of the (7 de julho de 2023). «FDA Converts Novel Alzheimer's Disease Treatment to Traditional Approval». FDA (em inglês). Consultado em 14 de fevereiro de 2024[ligação inativa]
- ↑ Francis RJ, Sharma SK, Springer C, Green AJ, Hope-Stone LD, Sena L, et al. (setembro de 2002). «A phase I trial of antibody directed enzyme prodrug therapy (ADEPT) in patients with advanced colorectal carcinoma or other CEA producing tumours». British Journal of Cancer. 87 (6): 600–607. PMC 2364249. PMID 12237768. doi:10.1038/sj.bjc.6600517
- ↑ Krauss WC, Park JW, Kirpotin DB, Hong K, Benz CC (2000). «Emerging antibody-based HER2 (ErbB-2/neu) therapeutics». Breast Disease. 11: 113–124. PMID 15687597. doi:10.3233/bd-1999-11110
- ↑ Joyce JA, Fearon DT (abril de 2015). «T cell exclusion, immune privilege, and the tumor microenvironment». Science. 348 (6230): 74–80. Bibcode:2015Sci...348...74J. PMID 25838376. doi:10.1126/science.aaa6204
- ↑ Hooks MA, Wade CS, Millikan WJ (1991). «Muromonab CD-3: a review of its pharmacology, pharmacokinetics, and clinical use in transplantation». Pharmacotherapy. 11 (1): 26–37. PMID 1902291. doi:10.1002/j.1875-9114.1991.tb03595.x
- ↑ Scolnik PA (2009). «mAbs: a business perspective». mAbs. 1 (2): 179–184. PMC 2725420. PMID 20061824. doi:10.4161/mabs.1.2.7736
- ↑ Kelley B (2009). «Industrialization of mAb production technology: the bioprocessing industry at a crossroads». mAbs. 1 (5): 443–452. PMC 2759494. PMID 20065641. doi:10.4161/mabs.1.5.9448
Links externos
- Manual de Gestão do Câncer: Princípios da Farmacoterapia Oncológica (inscrição necessária ) Arquivado em 2009-05-15 no Wayback Machine