Catálise enzimática
Catálise enzimática é o aumento da velocidade de um processo devido a uma biomolécula chamada "enzima". A maioria das enzimas são proteínas, e a maioria destes processos são reações químicas. Dentro da enzima, a catálise ocorre geralmente num sítio localizado chamado sítio ativo.
A maioria das enzimas é constituída predominantemente por proteínas e pode consistir numa única cadeia proteica ou em muitas cadeias no caso de um complexo multissubunitário. As enzimas incorporam frequentemente também componentes não proteicos, como iões metálicos ou moléculas orgânicas especializadas denominadas cofatores (por exemplo, trifosfato de adenosina). Muitos co-factores são vitaminas, e o seu papel como vitaminas está directamente ligado à sua utilização na catalisação de processos biológicos de metabolismo. A catálise das reações bioquímicas na célula é vital porque praticamente todas as reações metabólicas essenciais têm taxas muito baixas se não forem catalisadas. Um factor determinante da evolução das proteínas é a optimização destas actividades catalíticas, embora apenas as enzimas mais fundamentais operem perto dos limites da eficiência catalítica, e muitas enzimas estejam longe de ser óptimas. Fatores importantes na catálise enzimática são a catálise ácida e básica geral, a direção orbital, a restrição entrópica, os efeitos de orientação (i.e., catálise de chave e fechadura), bem como os efeitos de movimento inerentes à dinâmica das proteínas.[1]
Os mecanismos da catálise enzimática variam, mas são todos semelhantes, em princípio, a outros tipos de catálise química, pois o factor crucial é a redução da(s) barreira(s) de energia que separa(m) os reagentes (ou substratos) dos produtos. A redução da energia de activação (Ea) aumenta a fracção de moléculas de reagente que podem ultrapassar esta barreira e formar o produto. Um princípio importante é que, como apenas reduzem as barreiras energéticas entre produtos e reagentes, as enzimas catalisam sempre reações em ambos os sentidos e não podem impulsionar uma reação ou afetar a posição de equilíbrio, afetando apenas a velocidade com que esta é atingida. Tal como acontece com outros catalisadores, a enzima não é consumida ou alterada pela reação (como o substrato), mas sim reciclada, pelo que uma única enzima pode realizar várias rondas de catálise.
As enzimas são normalmente muito específicas, atuando apenas sobre determinados substratos. Algumas enzimas são absolutamente específicas, o que significa que apenas atuam sobre um substrato, enquanto outras apresentam especificidade de grupo e podem atuar sobre grupos químicos semelhantes, mas não idênticos, como a ligação peptídica em diferentes moléculas. Muitas enzimas têm especificidade estereoquímica, atuando sobre um estereoisómero, mas não sobre outro.[2]
Ajuste induzido
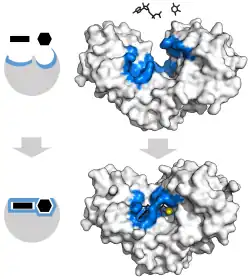
O modelo clássico da interação enzima-substrato é o modelo de ajuste induzido.[3] Este modelo propõe que a interação inicial entre a enzima e o substrato é relativamente fraca, mas que estas interações fracas induzem rapidamente mudanças conformacionais na enzima que fortalecem a ligação.
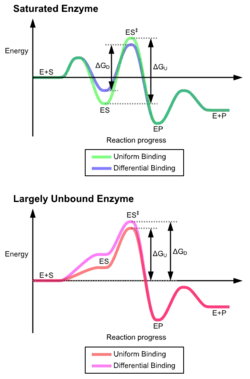
As vantagens do mecanismo de ajuste induzido devem-se ao efeito estabilizador da forte ligação da enzima. Existem dois mecanismos de ligação ao substrato: ligação uniforme, que apresenta uma forte ligação ao substrato, e ligação diferencial, que apresenta uma forte ligação ao estado de transição. O efeito estabilizador da ligação uniforme aumenta a afinidade tanto do substrato como do estado de transição, enquanto a ligação diferencial apenas aumenta a afinidade de ligação do estado de transição. As enzimas podem utilizar ambos e evoluíram para minimizar a energia de ativação da reação. As enzimas saturadas, isto é, com elevada afinidade de ligação ao substrato, requerem ligação diferencial para reduzir a energia de activação, enquanto as enzimas com substrato pequeno não ligado podem utilizar ligação diferencial ou ligação uniforme.[4]
Estes efeitos levaram a maioria das proteínas a utilizar o mecanismo de ligação diferencial para reduzir a energia de ativação, pelo que a maioria dos substratos apresenta uma elevada afinidade pela enzima enquanto se encontram no estado de transição. A ligação diferencial é realizada pelo mecanismo de ajuste induzido: o substrato liga-se primeiro fracamente, depois a enzima muda de conformação, aumentando a afinidade do estado de transição e estabilizando-o, reduzindo assim a energia de ativação necessária para o atingir.
No entanto, é importante esclarecer que o conceito de ajuste induzido não pode ser utilizado para racionalizar a catálise. Ou seja, a catálise química é definida como a redução de Ea‡ (quando o sistema já se encontra no ES‡) em relação ao Ea‡ da reação não catalisada em água (sem a enzima). O ajuste induzido apenas sugere que a barreira é menor na forma fechada, mas não nos diz nada sobre o motivo pelo qual ocorreu esta redução da barreira.
O ajuste induzido pode ser benéfico para a fidelidade do reconhecimento molecular na presença de competição e ruído através do mecanismo de revisão conformacional.[5]
Mecanismos de vias alternativas de reacção
Estas alterações conformacionais também aproximam os resíduos catalíticos das ligações químicas do sítio ativo dos substratos que serão alterados na reação. Após a ligação, um ou mais mecanismos de catálise reduzem a energia do estado de transição da reação, promovendo uma via química alternativa para a reação. Existem seis mecanismos possíveis de catálise "para além da barreira" e um mecanismo "através da barreira", que são os seguintes:
Proximidade e orientação
As interações enzima-substrato alinham os grupos químicos reativos e mantêm-nos próximos numa geometria ótima, o que aumenta a velocidade da reação. Isto reduz a entropia dos reagentes e, portanto, torna as reações de adição ou transferência menos desfavoráveis, daí a redução da entropia total quando os dois reagentes são convertidos num único produto. No entanto, este é um efeito geral e ocorre em reações sem adição ou transferência, onde isto ocorre devido a um aumento da "concentração efetiva" dos reagentes. Isto é compreensível quando se considera como os aumentos da concentração levam a um aumento da velocidade da reação: essencialmente, quando os reagentes estão mais concentrados, colidem com mais frequência e, por isso, reagem com mais frequência. Na catálise enzimática, a ligação dos reagentes à enzima restringe o espaço conformacional dos reagentes, mantendo-os na "orientação correta" e muito próximos uns dos outros, pelo que colidem mais frequentemente e, com a geometria correta, facilitam a reação desejada. A "concentração efectiva" é a concentração do reagente, livre em solução, que teria de existir para que este experimentasse a mesma frequência de colisão. Frequentemente, estas concentrações teóricas eficazes são fisicamente impossíveis e inatingíveis na prática, o que comprova o grande poder catalítico de muitas enzimas, que provocam um aumento massivo da velocidade em relação ao estado não catalisado.
| Por exemplo: |
| Reações semelhantes ocorrerão muito mais rapidamente se a reação for intermolecular. |
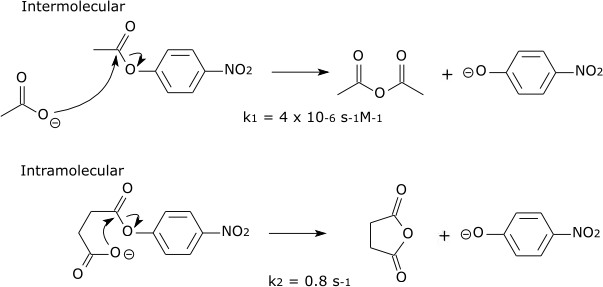
|
| A concentração efectiva de acetato na reacção intramolecular pode ser estimada como k2/k1 = 2 x 105 Molar. |
No entanto, a situação pode ser mais complexa, porque os estudos computacionais modernos estabeleceram que os exemplos tradicionais de efeitos de proximidade não podem ser directamente relacionados com efeitos entrópicos enzimáticos.[6][7][8] Além disso, verificou-se que a proposta entrópica original[9] sobrestimou muito a contribuição da entropia de orientação para a catálise.[10]
Dadores e aceitadores de protões
Os dadores e aceitadores de protões, ou seja, ácidos e bases, podem doar e aceitar protões para estabilizar o desenvolvimento de carga no estado de transição. Isto está relacionado com o princípio da catálise global, o da redução das barreiras energéticas, uma vez que os estados de transição gerais são estados de alta energia e, ao estabilizá-los, esta alta energia é reduzida, diminuindo a barreira. Uma característica fundamental da catálise enzimática, entre muitas catálises não biológicas, é que tanto a catálise ácida como a básica podem ser combinadas na mesma reação. Em muitos sistemas abióticos, os ácidos (grandes [H+]) ou as bases (grandes concentrações de H+ tornam-se mínimas, ou espécies com pares de eletrões) podem aumentar a velocidade da reação; mas, claro, o ambiente pode ter apenas um pH geral (medida de acidez ou basicidade (alcalinidade)). No entanto, como as enzimas são moléculas grandes, podem colocar grupos ácidos e básicos no seu sítio ativo para interagir com os seus substratos, e utilizam ambos os modos, independentemente do pH.
A catálise ácida ou básica geral é frequentemente utilizada para ativar nucleófilos e/ou grupos eletrofílicos, ou para estabilizar grupos de saída. Muitos aminoácidos com grupos ácidos e básicos são utilizados no sítio ativo, tais como ácido glutâmico, ácido aspártico, histidina, cisteína, tirosina, lisina e arginina, bem como serina e treonina. Além disso, o esqueleto molecular do peptídeo é frequentemente utilizado, com grupos carbonil e amida contendo N. A cistina e a histidina são as mais frequentemente implicadas, uma vez que ambas têm uma pKa próxima do pH neutro e, por isso, podem tanto aceitar como doar protões.
Muitos mecanismos de reacção que envolvem a catálise ácido-base envolvem um pKa substancialmente alterado. Esta alteração do pKa é possibilitada pelo ambiente local do resíduo.[carece de fontes].
| Condições | Ácidos | Bases |
|---|---|---|
| Ambiente hidrófobo | Incremento do pKa | Diminuição do pKa |
| Resíduos adjacentes de carga similar | Aumento do pKa | Diminuição do pKa |
| Ponte salina (e formação de ligações de hidrogénio) |
Diminuição do pKa | Aumento do pKa |
O pKa pode também ser significativamente influenciado pelo meio envolvente, ao ponto de os resíduos básicos em solução poderem atuar como doadores de protões e vice-versa.
| Por exemplo: |
| 'Tríade catalítica de uma serina-protease |
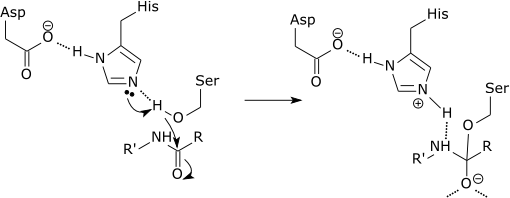
|
| A etapa inicial do mecanismo catalítico da serina-protease envolve a histidina do sítio activo aceitando um protão do resíduo de serina. Isto prepara a serina como um nucleófilo para atacar a ligação amida do substrato. Este mecanismo envolve a doação de um protão da serina (uma base, pKa 14) à histidina (um ácido, pKa 6), o que é possível devido ao ambiente local das bases. |
A modificação do pKa faz parte do mecanismo electrostático.[11] O efeito catalítico no exemplo acima está principalmente associado à redução do pKa do oxânion e ao aumento do pKa da histidina, enquanto a transferência de protões da serina para a histidina não é catalisada significativamente, uma vez que não é a barreira limitante da velocidade.[12] Note-se que, no exemplo apresentado, o ácido conjugado da histidina atua como um catalisador ácido global para a subsequente perda da amina de um intermediário tetraédrico. No entanto, as evidências que suportam este mecanismo proposto (Figura 4 da referência seguinte)[13] eram controversos.[14]
Catálise electrostática
A estabilização dos estados de transição carregados pode também ocorrer por resíduos do sítio ativo que formam ligações iónicas (ou interações de carga iónica parcial) com o intermediário. Estas ligações podem ser provenientes de cadeias laterais ácidas ou básicas encontradas em aminoácidos como a lisina, arginina, ácido aspártico ou ácido glutâmico ou de cofatores metálicos como o zinco. Os iões metálicos são particularmente eficazes e podem reduzir o pKa da água o suficiente para a tornar um nucleófilo eficaz.
Estudos sistemáticos de simulação computacional estabeleceram que os efeitos eletrostáticos fornecem, de longe, a maior contribuição para a catálise.[11] Isto pode incrementar a velocidade da reacción nun factor de ata 107.[15] Especificamente, foi demonstrado que a enzima proporciona um ambiente mais polar do que a água, e que os estados de transição iónicos são estabilizados por dipolos fixos. Isto é muito diferente da estabilização do estado de transição na água, onde as moléculas de água têm de pagar com uma "energia de reorganização"[16] para estabilizar os estados iónico e carregado. Assim, a catálise está associada ao facto de os grupos polares da enzima estarem pré-organizados.[17]
A magnitude do campo electrostático exercido pelo sítio activo da enzima nunca demonstrou estar altamente correlacionada com o aumento da taxa catalítica da enzima.[18]
A ligação ao substrato exclui geralmente a água do sítio ativo, reduzindo assim a constante dielétrica local para a de um solvente orgânico. Isto fortalece as interações eletrostáticas entre os substratos polares carregados e os sítios ativos. Além disso, a distribuição de carga nos sítios ativos está organizada de forma a estabilizar os estados de transição das reações catalisadas. Em diversas enzimas, estas distribuições de carga servem para guiar os substratos polares em direção aos seus locais de ligação, de modo a que as velocidades destas reações enzimáticas sejam superiores aos seus limites aparentes controlados por difusão.[carece de fontes]
| Por exemplo: |
| Mecanismo catalítico da carboxipeptidase |

|
| O intermediário tetraédrico é estabilizado por uma ligação iónica parcial entre o ião Zn2+ e a carga negativa no oxigénio. |
Catálise covalente
A catálise covalente envolve a formação de uma ligação covalente transitória pelo substrato com resíduos no sítio ativo da enzima ou com um co-fator. Isto adiciona um intermediário covalente adicional à reação e ajuda a reduzir a energia dos estados de transição posteriores da reação. A ligação covalente deve ser quebrada numa fase posterior da reação para regenerar a enzima. Este mecanismo é utilizado pela tríade catalítica de enzimas como as proteases, como a quimotripsina e a tripsina, nas quais se formou um intermediário acil-enzima. Um mecanismo alternativo é a formação de uma base de Schiff utilizando a amina livre de um resíduo de lisina, como observado na enzima aldolase durante a glicólise.
Algumas enzimas utilizam co-factores não aminoácidos, como o fosfato de piridoxal (PLP) ou o pirofosfato de tiamina (TPP) para formar intermediários covalentes com moléculas reagentes.[19][20] Estes intermediários covalentes funcionam reduzindo a energia dos estados de transição subsequentes, de forma semelhante à estabilização proporcionada pelos intermediários covalentes formados com resíduos de aminoácidos do sítio activo. No entanto, as capacidades dos co-factores permitem que as enzimas realizem reacções que os resíduos de aminoácidos secundários por si só não conseguiriam. As enzimas que utilizam estes co-factores incluem a enzima dependente de PLP aspartato transaminase e a enzima dependente de TPP piruvato desidrogenase..[21][22]
Em vez de reduzir a energia de ativação de uma via de reação, a catálise covalente fornece uma via alternativa para a reação (através de um intermediário covalente) e, portanto, é distinta da catálise verdadeira. Por exemplo, a energética da ligação covalente a uma molécula de serina na quimotripsina deve ser comparada com a conhecida ligação covalente ao nucleófilo numa reacção em solução não catalisada. Uma proposta verdadeira para a catálise covalente (em que a barreira é mais pequena do que a barreira correspondente em solução) exigiria, por exemplo, uma ligação covalente parcial ao estado de transição por um grupo da enzima (por exemplo, uma ligação de hidrogénio muito forte), e tais efeitos não contribuem significativamente para a catálise.
Catálise de Iões Metálicos
Um ião metálico no sítio ativo participa na catálise coordenando a estabilização da carga e a blindagem. Devido à carga positiva do metal, apenas as cargas negativas podem ser estabilizadas por iões metálicos.[23] No entanto, os iões metálicos são vantajosos na catálise biológica porque não são afetados pelas alterações do pH.[24] Os iões metálicos podem também atuar ionizando a água, funcionando como um ácido de Lewis.[25] Os iões metálicos podem também ser agentes de oxidação e redução.[26]
Tensão de ligação
Este é o principal efeito da ligação por ajuste induzida, em que a afinidade da enzima pelo estado de transição é maior do que a do próprio substrato. Isto induz redistribuições estruturais na molécula que tensionam as ligações do substrato para mais perto da conformação do estado de transição, reduzindo assim a diferença de energia entre o substrato e o estado de transição e ajudando a catalisar a reação.
No entanto, o efeito de tensão é, na realidade, mais um efeito desestabilizador do estado fundamental do que um efeito estabilizador do estado de transição.[11][27] Por outro lado, as enzimas são muito flexíveis e não podem aplicar um grande efeito de tensão.[28]
Para além da tensão de ligação exercida sobre o substrato, esta também pode ser induzida na própria enzima para activar resíduos do sítio activo.
| Por exemplo: |
| Conformações do substrato, substrato ligado e estado de transição da enzima lisozima. |
| O substrato, ao ligar-se, é distorcido da sua conformação de meia cadeira do anel hexose (devido ao impedimento estérico com aminoácidos na proteína que obrigam o C6 equatorial a estar na posição axial) para a conformação de cadeira,[29] que tem um formato semelhante ao estado de transição. |
Tunelamento quântico
Estes mecanismos tradicionais de "atravessar a barreira" foram questionados em alguns casos por modelos e observações de mecanismos de "através da barreira" (o tunelamento quântico). Algumas enzimas operam com cinética mais rápida do que seria previsto pelo ΔG‡ clássico. Nos modelos de "através da barreira", um electrão ou um protão podem atravessar barreiras de activação.[30][31] O tunelamento quântico de protões foi observado na oxidação da triptamina por uma amina desidrogenase aromática.[32]
O tunelamento quântico não parece proporcionar uma vantagem catalítica significativa, uma vez que as contribuições do tunelamento são semelhantes nas reações catalisadas e não catalisadas em solução.[31][33][34][35] No entanto, a contribuição do tunelamento (tipicamente aumentando as constantes de velocidade por um factor de ~1000[32] em comparação com a velocidade das reacções na via clássica "através da barreira") é provavelmente crucial para a viabilidade dos organismos biológicos. Isto realça a importância geral das reações de tunelamento na biologia.
Em 1971-1972, foi formulado um dos primeiros modelos mecânicos quânticos de catálise enzimática.[36][37]
Enzima ativa
A energia de ligação do complexo enzima-substrato não pode ser considerada uma energia externa necessária para a activação do substrato. O elevado conteúdo energético da enzima pode ser transferido primeiramente para algum grupo energético específico X1 do sítio catalítico da enzima para o sítio final do primeiro reagente ligado. De seguida, outro grupo X2 deve ser transferido do segundo reagente (ou do segundo grupo do reagente único) para o sítio activo para completar a conversão do substrato em produto e a regeneração da enzima.[38]
Podemos apresentar a reação enzimática completa como duas reações acopladas:
-
S
1 + EX
1 → S
1EX
1 → P
1 + EP
2(1)
-
S
2 + EP
2 → S
2EP
2 → P
2 + EX
2(2)
Pode observar-se na reação (1) que o grupo X1 da enzima ativa aparece no produto devido à possibilidade de uma reação de troca dentro da enzima para evitar a inibição eletrostática e a repulsão atómica. Assim, representamos a enzima ativa como um potente reagente da reação enzimática. A reação (2) mostra a conversão incompleta do substrato porque o seu grupo X2 permanece dentro da enzima. Esta abordagem foi proposta anteriormente como uma ideia que dependia de conversões enzimáticas hipoteticamente extremamente elevadas (enzima cataliticamente perfeita).[39]
O ponto essencial para a verificação da presente abordagem é que o catalisador deve ser um complexo da enzima com o grupo de transferência da reacção. Este aspeto químico é suportado pelos mecanismos bem estudados das diversas reações enzimáticas. Considere-se a hidrólise da ligação peptídica catalisada por uma proteína pura como a α-quimotripsina (uma enzima que actua sem co-factor), que é um membro bem estudado da família das serina-proteases, ver.[40]
Apresentamos os resultados experimentais desta reação em duas etapas químicas:
-
S
1 + EH → P
1 + EP
2(3)
-
EP
2 + H–O–H → EH + P
2(4)
em que S1 é um polipeptídeo, P1 e P2 são produtos. A primeira etapa química (3) envolve a formação de um intermediário acil-enzima covalente. A segunda etapa (4) é a etapa de desacilação. É importante salientar que o grupo H+, inicialmente encontrado na enzima, mas não na água, surge no produto antes da etapa de hidrólise; pode, portanto, ser considerado um grupo adicional da reação enzimática.
Assim, a reação (3) mostra que a enzima atua como um reagente potente na reação. De acordo com o conceito proposto, o transporte de H da enzima promove a primeira conversão do reagente, a quebra da primeira ligação química inicial (entre os grupos P1 e P2). A etapa de hidrólise provoca a quebra da segunda ligação química e a regeneração da enzima.
Os mecanismos químicos propostos não dependem da concentração dos substratos ou dos produtos no meio. No entanto, uma alteração nas suas concentrações provoca principalmente alterações na energia livre na primeira e última etapas das reações (1) e (2), devido a alterações no conteúdo de energia livre de cada molécula, seja S ou P, em solução aquosa. Esta estratégia é consistente com o seguinte mecanismo de contração muscular. A etapa final da hidrólise do ATP no músculo esquelético é a libertação do produto provocada pela associação das cabeças de miosina com a actina.[41] O fecho da fenda de ligação da actina durante a reação de associação está estruturalmente acoplado com a abertura da bolsa de ligação do nucleótido no sítio ativo da miosina.[42]
As etapas finais da hidrólise do ATP incluem a libertação rápida de fosfato e a libertação lenta de ADP.[43][44] A libertação de um anão fosfato do anão ADP ligado em solução aquosa pode ser considerada uma reação exergónica, uma vez que o anão fosfato apresenta uma baixa massa molecular.
Assim, concluímos que a libertação primária do fosfato inorgânico H2PO4− provoca a transformação de uma parte significativa da energia livre da hidrólise do ATP na energia cinética do fosfato solvatado, produzindo fluxo activo (fluxo de subestruturas celulares provocado pela miosina e actina como na ciclose, neste caso, o deslizamento dos filamentos na fibra muscular). Esta suposição de uma transdução mecanoquímica local está de acordo com o mecanismo de contração muscular de Tirosh, no qual a força muscular deriva de uma ação integrada de fluxo ativo criado pela hidrólise do ATP.[45][46]
Exemplos de mecanismos catalíticos
Na realidade, a maioria dos mecanismos enzimáticos envolve uma combinação de vários tipos de catálise.
Triose-fosfato isomerase
A Triose-fosfato isomerase (EC 5.3.1.1) catalisa a interconversão reversível dos dois isómeros de triose fosfato di-hidroxiacetona fosfato e D-gliceraldeído 3-fosfato. O mecanismo catalítico envolve a formação de um intermediário "enediol".
Tripsina
Tripsina (EC 3.4.21.4) é uma serina-protease que cliva substratos proteicos após resíduos de lisina ou arginina, utilizando uma tríade catalítica para realizar a catálise covalente e um buraco de oxânion para estabilizar a acumulação de carga nos estados de transição.
Aldolase
A [aldolase]] (ou aldolase de frutose-bisfosfato EC 4.1.2.13) catalisa a clivagem de uma frutose 1,6-bisfosfato (F-1,6-BP) em gliceraldeído 3-fosfato e di-hidroxiacetona fosfato (DHAP). Forma um intermediário protonado base de Schiff ligado a uma lisina altamente conservada no sítio activo, no carbono carbonílico do DHAP. Além disso, os resíduos de tirosina são cruciais neste mecanismo, uma vez que atuam como aceptores de hidrogénio estabilizadores.
Difusividade enzimática
O aparecimento de estudos com uma única molécula na década de 2010 levou à observação de que o movimento das enzimas não ligadas aumenta com o aumento da concentração do substrato e aumenta a entalpia de reação.[47] Observações subsequentes sugerem que este aumento da difusividade é causado pela alteração transitória do centro de massa da enzima, resultando num "efeito de recuo propulsor da enzima".[48]
Semelhança de reação
A semelhança entre reações enzimáticas ([1]) pode ser calculada utilizando dados de alterações de ligação, centros de reação ou métricas de subestrutura ([2] Arquivado em 2019-05-30 no Wayback Machine).[49]
Referências
- ↑ Kamerlin SC, Warshel A (Maio de 2010). «No dealbar do século XXI: será a dinâmica o elo perdido para a compreensão da catálise enzimática?» 6 ed. Proteínas. 78: 1339–1375. PMC 2841229
 . PMID 20099310. doi:10.1002/prot.22654
. PMID 20099310. doi:10.1002/prot.22654
- ↑ Laidler KJ (1978). Físico-Química com Aplicações Biológicas. [S.l.]: Benjamin/Cummings. p. 427. ISBN 978-0-8053-5680-9
- ↑ Koshland DE (fevereiro de 1958). «Application of a Theory of Enzyme Specificity to Protein Synthesis». Proceedings of the National Academy of Sciences of the United States of America. 44 (2): 98–104. Bibcode:1958PNAS...44...98K. PMC 335371. PMID 16590179. doi:10.1073/pnas.44.2.98

- ↑ Anslyn EV, Dougherty DA (2006). Modern Physical Organic Chemistry. [S.l.]: University Science Books. ISBN 978-1-891389-31-3
- ↑ Savir Y, Tlusty T (Maio de 2007). Scalas E, ed. «Revisão conformacional: o impacto das alterações conformacionais na especificidade do reconhecimento molecular» 5 ed. PLOS ONE. 2: e468. Bibcode:2007PLoSO...2..468S. PMC 1868595. PMID 17520027. doi:10.1371/journal.pone.0000468
- ↑ Stanton RV, Peräkylä M, Bakowies D, Kollman PA (1998). «Combined ab initio and Free Energy Calculations To Study Reactions in Enzymes and Solution: Amide Hydrolysis in Trypsin and Aqueous Solution». J. Am. Chem. Soc. 120 (14): 3448–3457. doi:10.1021/ja972723x
- ↑ Kuhn B, Kollman PA (2000). «QM-FE and Molecular Dynamics Calculations on Catechol O-Methyltransferase: Free Energy of Activation in the Enzyme and in Aqueous Solution and Regioselectivity of the Enzyme-Catalyzed Reaction». J. Am. Chem. Soc. 122 (11): 2586–2596. doi:10.1021/ja992218v
- ↑ Bruice TC, Lightstone FC (1999). «Ground State and Transition State Contributions to the Rates of Intramolecular and Enzymatic Reactions». Acc. Chem. Res. 32 (2): 127–136. doi:10.1021/ar960131y
- ↑ Page MI, Jencks WP (agosto de 1971). «Entropic contributions to rate accelerations in enzymic and intramolecular reactions and the chelate effect». Proceedings of the National Academy of Sciences of the United States of America. 68 (8): 1678–1683. Bibcode:1971PNAS...68.1678P. PMC 389269. PMID 5288752. doi:10.1073/pnas.68.8.1678
- ↑ Warshel A, Parson WW (novembro de 2001). «Dynamics of biochemical and biophysical reactions: insight from computer simulations». Quarterly Reviews of Biophysics. 34 (4): 563–679. PMID 11852595. doi:10.1017/s0033583501003730
- ↑ a b c Warshel A, Sharma PK, Kato M, Xiang Y, Liu H, Olsson MH (agosto de 2006). «Electrostatic basis for enzyme catalysis». Chemical Reviews. 106 (8): 3210–3235. PMID 16895325. doi:10.1021/cr0503106
- ↑ Warshel A, Naray-Szabo G, Sussman F, Hwang JK (maio de 1989). «How do serine proteases really work?». Biochemistry. 28 (9): 3629–3637. PMID 2665806. doi:10.1021/bi00435a001
- ↑ Fersht AR, Requena Y (dezembro de 1971). «Mechanism of the -chymotrypsin-catalyzed hydrolysis of amides. pH dependence of kc and Km . Kinetic detection of an intermediate». Journal of the American Chemical Society. 93 (25): 7079–7087. PMID 5133099. doi:10.1021/ja00754a066
- ↑ Zeeberg B, Caswell M, Caplow M (abril de 1973). «Concerning a reported change in rate-determining step in chymotrypsin catalysis». Journal of the American Chemical Society. 95 (8): 2734–2735. PMID 4694533. doi:10.1021/ja00789a081
- ↑ Voet D, Voet JG (2011). Biochemistry. [S.l.]: John Wiley & Sons. OCLC 808679090
- ↑ Marcus RA (1965). «On the Theory of Electron-Transfer Reactions. VI. Unified Treatment for Homogeneous and Electrode Reactions» (PDF). J. Chem. Phys. 43 (2): 679–701. Bibcode:1965JChPh..43..679M. doi:10.1063/1.1696792
- ↑ Warshel A (novembro de 1978). «Energetics of enzyme catalysis». Proceedings of the National Academy of Sciences of the United States of America. 75 (11): 5250–5254. Bibcode:1978PNAS...75.5250W. PMC 392938. PMID 281676. doi:10.1073/pnas.75.11.5250
- ↑ Fried SD, Bagchi S, Boxer SG (dezembro de 2014). «Extreme electric fields power catalysis in the active site of ketosteroid isomerase». Nova York, N.Y. Science. 346 (6216): 1510–4. Bibcode:2014Sci...346.1510F. PMC 4668018. PMID 25525245. doi:10.1126/science.1259802
- ↑ Toney, M. D. "Reaction specificity in pyridoxal enzymes." Archives of biochemistry and biophysics (2005) 433: 279-287
- ↑ «Micronutrient Information Center, Oregon State University». Consultado em 30 de setembro de 2009. Cópia arquivada em 21 de março de 2015
- ↑ Voet D, Voet JG (2004). Biochemistry. [S.l.]: John Wiley & Sons Inc. pp. 986–989. ISBN 978-0-471-25090-6
- ↑ Voet D, Voet JG (2004). Biochemistry. [S.l.]: John Wiley & Sons Inc. pp. 604–606. ISBN 978-0-471-25090-6
- ↑ Piccirilli JA, Vyle JS, Caruthers MH, Cech TR (janeiro de 1993). «Metal ion catalysis in the Tetrahymena ribozyme reaction». Nature. 361 (6407): 85–88. Bibcode:1993Natur.361...85P. PMID 8421499. doi:10.1038/361085a0
- ↑ Bender ML (1 de janeiro de 1962). «Metal Ion Catalysis of Nucleophilic Organic Reactions in Solution». Reactions of Coordinated Ligands. Col: Advances in Chemistry. 37. [S.l.]: American Chemical Society. pp. 19–36. ISBN 978-0-8412-0038-8. doi:10.1021/ba-1963-0037.ch002
- ↑ Fife TH, Przystas TJ (1 de fevereiro de 1985). «Divalent metal ion catalysis in the hydrolysis of esters of picolinic acid. Metal ion promoted hydroxide ion and water catalyzed reactions». Journal of the American Chemical Society. 107 (4): 1041–1047. ISSN 0002-7863. doi:10.1021/ja00290a048
- ↑ Stadtman ER (1 de janeiro de 1990). «Metal ion-catalyzed oxidation of proteins: biochemical mechanism and biological consequences». Free Radical Biology & Medicine. 9 (4): 315–325. PMID 2283087. doi:10.1016/0891-5849(90)90006-5
- ↑ Jencks WP (1987) [1969]. Catalysis in Chemistry and Enzymology. Col: McGraw-Hill series in advanced chemistry reprint ed. Nova York: Dover Publications. ISBN 978-0-486-65460-7
- ↑ Warshel A, Levitt M (maio de 1976). «Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme». Journal of Molecular Biology. 103 (2): 227–249. PMID 985660. doi:10.1016/0022-2836(76)90311-9
- ↑ Voet D, Voet JG, Pratt CW (2013). Fundamentals of Biochemistry: Life at the Molecular Level 4ª ed. Hoboken, NJ: Wiley. ISBN 978-0-470-54784-7
- ↑ Garcia-Viloca M, Gao J, Karplus M, Truhlar DG (janeiro de 2004). «How enzymes work: analysis by modern rate theory and computer simulations». Science. 303 (5655): 186–195. Bibcode:2004Sci...303..186G. PMID 14716003. doi:10.1126/science.1088172
- ↑ a b Olsson MH, Siegbahn PE, Warshel A (março de 2004). «Simulations of the large kinetic isotope effect and the temperature dependence of the hydrogen atom transfer in lipoxygenase». Journal of the American Chemical Society. 126 (9): 2820–2828. PMID 14995199. doi:10.1021/ja037233l
- ↑ a b Masgrau L, Roujeinikova A, Johannissen LO, Hothi P, Basran J, Ranaghan KE, et al. (abril de 2006). «Atomic description of an enzyme reaction dominated by proton tunneling». Science. 312 (5771): 237–241. Bibcode:2006Sci...312..237M. PMID 16614214. doi:10.1126/science.1126002
- ↑ Hwang JK, Warshel A (1996). «How important are quantum mechanical nuclear motions in enzyme catalysis». J. Am. Chem. Soc. 118 (47): 11745–11751. doi:10.1021/ja962007f
- ↑ Ball P (setembro de 2004). «Enzymes: by chance, or by design?». Nature. 431 (7007): 396–397. Bibcode:2004Natur.431..396B. PMID 15385982. doi:10.1038/431396a
- ↑ Olsson MH, Parson WW, Warshel A (maio de 2006). «Dynamical contributions to enzyme catalysis: critical tests of a popular hypothesis». Chemical Reviews. 106 (5): 1737–1756. PMID 16683752. doi:10.1021/cr040427e
- ↑ Vol'kenshtein MV, Dogonadze RR, Madumarov AK, Urushadze ZD, Kharkats YI (1972). «The theory of enzyme catalysis». Moscow. Molecular Biology. 6 (3): 347–353. PMID 4645409
- ↑ Volkenshtein MV, Dogonadze RR, Madumarov AK, Urushadze ZD, Kharkats Yu I (1973). «Electronic and Conformational Interactions in Enzyme Catalysis.». Konformatsionnie Izmenenia Biopolimerov v Rastvorakh. Moscow: Nauka Publishing House. pp. 153–157
- ↑ Foigel AG (julho de 2011). «Is the enzyme a powerful reactant of the biochemical reaction?». Molecular and Cellular Biochemistry. 352 (1–2): 87–89. PMID 21318350. doi:10.1007/s11010-011-0742-4
- ↑ Fogel AG (Agosto de 1982). «Cooperatividade das reações enzimáticas e aspetos moleculares da transdução de energia» 1 ed. Molecular and Cellular Biochemistry. 47: 59–64. PMID 7132966. doi:10.1007/bf00241567
- ↑ Hengge AC, Stein RL (Janeiro de 2004). «Papel da mobilidade conformacional das proteínas na catálise enzimática: acilação da alfa-quimotripsina por substratos peptídicos específicos» 3 ed. Bioquímica. 43: 742–747. PMID 14730979. doi:10.1021/bi030222k
- ↑ Lymn RW, Taylor EW (dezembro de 1971). «Mechanism of adenosine triphosphate hydrolysis by actomyosin». Biochemistry. 10 (25): 4617–4624. PMID 4258719. doi:10.1021/bi00801a004
- ↑ Holmes KC, Angert I, Kull FJ, Jahn W, Schröder RR (setembro de 2003). «Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide». Nature. 425 (6956): 423–427. Bibcode:2003Natur.425..423H. PMID 14508495. doi:10.1038/nature02005
- ↑ Siemankowski RF, Wiseman MO, White HD (fevereiro de 1985). «ADP dissociation from actomyosin subfragment 1 is sufficiently slow to limit the unloaded shortening velocity in vertebrate muscle». Proceedings of the National Academy of Sciences of the United States of America. 82 (3): 658–662. Bibcode:1985PNAS...82..658S. PMC 397104. PMID 3871943. doi:10.1073/pnas.82.3.658
- ↑ White HD, Belknap B, Webb MR (setembro de 1997). «Kinetics of nucleoside triphosphate cleavage and phosphate release steps by associated rabbit skeletal actomyosin, measured using a novel fluorescent probe for phosphate». Biochemistry. 36 (39): 11828–11836. PMID 9305974. doi:10.1021/bi970540h
- ↑ Tirosh R, Low WZ, Oplatka A (março de 1990). «Movimento de translação dos filamentos de actina na presença de meromiosina pesada e MgATP, medido pelo alargamento Doppler do espalhamento de luz laser». Biochimica et Biophysica Acta (BBA) - Estrutura das Proteínas e Enzimologia Molecular. 1037 (3): 274–280. PMID 2178685. doi:10.1016/0167-4838(90)90025-b
- ↑ Tirosh R (2006). «Protões balísticos e soluções aquosas induzidas por micro-ondas (solitons) em transformações bioenergéticas» 9 ed. Int. J. Mol. Sci. 7: 320–345. doi:10.3390/i7090320
- ↑ Muddana HS, Sengupta S, Mallouk TE, Sen A, Butler PJ (fevereiro de 2010). «Substrate catalysis enhances single-enzyme diffusion». Journal of the American Chemical Society. 132 (7): 2110–2111. PMC 2832858. PMID 20108965. doi:10.1021/ja908773a
- ↑ Riedel C, Gabizon R, Wilson CA, Hamadani K, Tsekouras K, Marqusee S, et al. (janeiro de 2015). «The heat released during catalytic turnover enhances the diffusion of an enzyme». Nature. 517 (7533): 227–230. Bibcode:2015Natur.517..227R. PMC 4363105. PMID 25487146. doi:10.1038/nature14043
- ↑ Rahman SA, Cuesta SM, Furnham N, Holliday GL, Thornton JM (fevereiro de 2014). «EC-BLAST: a tool to automatically search and compare enzyme reactions». Nature Methods. 11 (2): 171–174. PMC 4122987. PMID 24412978. doi:10.1038/nmeth.2803