Autofagia
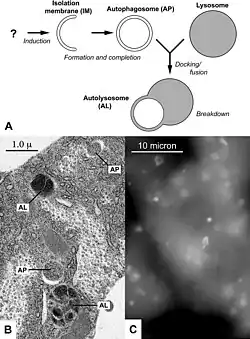
A autofagia (ou autofagocitose) é um processo catabólico celular que dá origem à degradação de componentes da própria célula utilizando os lisossomas. É um processo estreitamente regulado que desempenha uma função normal no crescimento celular, diferenciação, e na homeostase, e ajuda a manter um equilíbrio entre a síntese, a degradação e o reciclado dos produtos celulares. Consiste num dos principais mecanismos por meio dos quais uma célula em estado de desnutrição redistribui os nutrientes dos processos menos necessários aos essenciais.
A célula elimina organelas envelhecidas utilizando este mesmo mecanismo, que inclui a formação de vesículas com o auxílio do retículo endoplasmático liso: o organelo obsoleto é envolto numa membrana derivada desse mesmo retículo formando-se o chamado autofagossoma. De seguida, o autofagossoma, seguindo o mesmo caminho dos fagossomas, funde-se com um endossoma secundário, recebendo enzimas hidrolíticas do Complexo de Golgi. É, deste modo, transformado em fagolisossoma. O processo culmina com a degradação do organelo pela acção das enzimas.
Em células como os neurónios, hepatócitos e células musculares cardíacas, os fagolisossomas não completam a digestão total do organelo, sendo convertidos em corpos residuais. Com o avanço da idade, esses corpos formam pigmentos de inclusão que são acumulados no citosol. A autofagia pode ser estimulada em determinadas situações, como, por exemplo, durante o jejum prolongado, aparecendo numerosos autofagossomas nos hepatócitos com o objetivo de converter os componentes da célula em alimento para prolongar a sobrevivência do organismo.
O biólogo pesquisador Japonês Yoshinori Ohsumi foi premiado em 2016 com o Prémio Nobel da Medicina pela sua investigação relativa à autofagia.
História
Reconhecida nos anos 1970 por Daniel Klionsky, pesquisador da Universidade de Michigan, Estados Unidos, a autofagia passou quase três décadas vista apenas como uma forma, inicialmente sem muita importância, de a célula se livrar de si mesma. Por essa razão, foi chamada de morte celular programada tipo 2 para diferenciar da apoptose, ou morte tipo 1, muito mais estudada. Pode-se dizer que a autofagia antecede a morte celular ou que é cruzada à morte celular, mas hoje não é mais correto afirmar que a autofagia seja um tipo de morte celular, pois sua existência está melhor associada a resistência e sobrevivência.
Os genes que controlam a autofagia começaram a ser identificados em 1997, inicialmente em leveduras, organismos unicelulares, empregados na fabricação de pão, vinho, cerveja e álcool combustível. A partir dos genes, os especialistas conheceram quais são e como interagem as proteínas que levam adiante esse mecanismo flexível de reciclagem de componentes celulares.
O biólogo pesquisador Japonês Yoshinori Ohsumi foi premiado em 2016 com o Prêmio Nobel da Medicina pela sua investigação relativa à autofagia.
Mecanismos moleculares
Foram identificadas algumas dezenas de proteínas diferentes em células de levedura e de animais que participam no processo, que deve ser regulado rigorosamente: um pouco a mais ou um pouco a menos já pode ser deletério. Todo o processo ocorre na seguinte sequência de etapas:
- Indução por ativação de moléculas sinalizadoras: proteínas-cinase (incluindo o complexo mTOR 1) que retransmitem informação sobre a condição metabólica da célula se tornam ativadas e sinalizam para a maquinaria autofágica.
- Nucleação e expansão de uma membrana delimitante em forma de crescente: Vesículas de membrana, caracterizadas pela presença de ATG9, a única proteína transmembrana envolvida no processo, são recrutadas para um sítio de montagem, onde elas concentram a formação do autofagossomo (ou vacúolo autofágico). A ATG9 não é incorporada no autofagossomo: uma via de recuperação deve removê-la da estrutura de montagem.
- Fechamento da membrana ao redor do alvo para formar um autofagossomo delimitado por dupla membrana selado.
- Fusão do autofagossomo com lisossomos, catalisada pelas SNAREs, e formação do fagolisossomo.
- Digestão da membrana interna e dos conteúdos do lúmen do autofagossomo.
Em células como os neurônios, hepatócitos e células musculares cardíacas, os fagolisossomos não completam a digestão total da organela, sendo convertidos em corpos residuais. Com o avanço da idade, esses corpos formam pigmentos de inclusão que são acumulados no citosol.
Tipos de autofagia
A autofagia pode ser tanto não seletiva como seletiva. Na autofagia não seletiva, uma porção do citoplasma é sequestrada em autofagossomos. Pode ocorrer, por exemplo, em condições de privação alimentar: quando os nutrientes externos são limitados (jejum prolongado), os metabólitos derivados da digestão do citosol capturado podem ajudar a célula a sobreviver. Na autofagia seletiva, cargas específicas são empacotadas dentro dos autofagossomos que tendem a conter pouco citosol, e sua forma reflete a forma da carga. A autofagia seletiva medeia a degradação de mitocôndrias, ribossomos e RE que estão debilitados ou indesejados; ela também pode ser utilizada para destruir micróbios invasores.
A autofagia seletiva de mitocôndrias deterioradas ou danificadas é chamada de mitofagia. Quando as mitocôndrias funcionam normalmente, a membrana interna mitocondrial é energizada por um gradiente eletroquímico de H+ que direciona a síntese de ATP e a importação de proteínas precursoras mitocondriais e de metabólitos. As mitocôndrias danificadas não podem manter o gradiente, então a importação de proteínas é bloqueada. Como consequência, uma proteína-cinase chamada Pink1, que, em geral, é importada para as mitocôndrias, fica retida sobre a superfície mitocondrial onde recruta a ubiquitina-ligase Parkin do citosol. A Parkin realiza a ubiquitinação das proteínas da membrana mitocondrial externa, o que marca a organela para destruição seletiva nos autofagossomos. Mutações na Pink1 ou Parkin causam uma forma de aparecimento precoce da doença de Parkinson, uma doença degenerativa do sistema nervoso central. Não se sabe por que os neurônios que morrem prematuramente nessa doença são particularmente dependentes da mitofagia.
Biologia molecular
ATG é a abreviatura de "AuTophaGy" (autofagia em inglês), que se aplica tanto a genes como a proteínas relacionadas com o processo biológico da autofagia.[1] Existem cerca de 16 a 20 genes ATG conservados que codificam muitas proteínas ATG essenciais conservadas desde leveduras até seres humanos.[1] ATG pode fazer parte do nome da proteína (como ATG7 [en]) ou do nome do gene (como ATG7),[2] embora nem todas as proteínas e genes ATG sigam esse padrão (como ULK1).[1]
Para dar exemplos específicos, a enzima UKL1 (complexo quinase) induz a biogénese do autofagossoma, e a ATG13 (proteína 13 relacionada com a autofagia) é necessária para a formação do fagossoma.[3]
A autofagia é executada pelos genes ATG. Antes de 2003, eram utilizados dez ou mais nomes, mas depois dessa data, uma nomenclatura unificada foi criada por pesquisadores da autofagia fúngica.[4] Os primeiros genes da autofagia foram identificados por triagens genéticas realizadas em Saccharomyces cerevisiae.[5][6][7][8][9] Após a sua identificação, esses genes foram caracterizados funcionalmente e os seus ortólogos em uma variedade de organismos diferentes foram identificados e estudados.[10][11] Atualmente, trinta e seis proteínas Atg foram classificadas como especialmente importantes para a autofagia, das quais 18 pertencem ao mecanismo central.[12]
Nos mamíferos, a deteção de aminoácidos e sinais adicionais, como fatores de crescimento e espécies reativas de oxigénio, regulam a atividade das proteínas cinases mTOR e AMPK.[11][13] Estas duas cinases regulam a autofagia através da fosforilação inibitória das cinases ULK1 e ULK2 semelhantes à Unc-51 (homólogas mamíferas da Atg1).[14] A indução da autofagia resulta na desfosforilação e ativação das proteínas quinases ULK. A ULK faz parte de um complexo proteico que contém Atg13, Atg101 [en] e FIP200. A ULK fosforila e ativa a Beclin-1 (homóloga mamífera da Atg6),[15] que também faz parte de um complexo proteico. O complexo Beclin-1 induzível por autofagia[16] contém as proteínas PIK3R4 [en](p150), Atg14L e a fosfatidilinositol 3-fosfato quinase (PI(3)K) Vps34 [en] de classe III.[17] Os complexos ativos ULK e Beclin-1 se relocalizam no local de início do autofagossomo, o fagóforo, onde ambos contribuem para a ativação dos componentes da autofagia a jusante.[18][19]
Uma vez ativado, o VPS34 fosforila o fosfatidilinositol lipídico para gerar fosfatidilinositol 3-fosfato (PtdIns(3)P) na superfície do fagóforo. O PtdIns(3)P gerado é usado como ponto de acoplamento para proteínas que abrigam um motivo de ligação PtdIns(3)P. Recentemente, demonstrou-se que a WIPI2 [en], uma proteína de ligação ao PtdIns(3)P da família de proteínas WIPI (proteína de repetição WD que interage com fosfoinositídeos), se liga fisicamente à ATG16L1.[20] A Atg16L1 é um membro de um complexo proteico semelhante ao E3 envolvido num dos dois sistemas de conjugação semelhantes à ubiquitina essenciais para a formação de autofagossomas. As membranas derivadas do cis-Golgi FIP200 fundem-se com as membranas endossomais positivas para ATG16L1 para formar o profagóforo denominado HyPAS (estrutura pré-autofagossomal híbrida).[21] A ligação de ATG16L1 a WIPI2[22] medeia a atividade de ATG16L1. Isto leva à conversão a jusante do profagóforo em fagóforo ATG8-positivo[21] através de um sistema de conjugação semelhante à ubiquitina.
O primeiro dos dois sistemas de conjugação semelhantes à ubiquitina envolvidos na autofagia liga covalentemente a proteína semelhante à ubiquitina Atg12 à Atg5. A proteína conjugada resultante liga-se então à ATG16L1 para formar um complexo semelhante ao E3, que funciona como parte do segundo sistema de conjugação semelhante à ubiquitina.[23] Este complexo liga-se e ativa a Atg3 [en], que liga covalentemente os homólogos mamíferos da proteína ATG8 [en] (LC3A-C [en], GATE16 e GABARAPL1-3) da levedura semelhante à ubiquitina, sendo as proteínas LC3 as mais estudadas, ao fosfatidiletanolamina (PE) lipídico na superfície dos autofagossomas.[24] O LC3 lipídico contribui para o fechamento dos autofagossomas[25] e permite a ligação de cargas específicas e proteínas adaptadoras, como Sequestosome-1/p62 [en].[26] O autofagossoma completo então se funde com um lisossoma por meio da ação de várias proteínas, incluindo SNAREs[27][28] e UVRAG [en].[29] Após a fusão, o LC3 é retido no lado interno da vesícula e degradado junto com a carga, enquanto as moléculas de LC3 ligadas ao lado externo são clivadas pelo Atg4 e recicladas.[30] O conteúdo do autolisossoma é subsequentemente degradado e seus blocos de construção são liberados da vesícula através da ação de permeases.[31]
A sirtuína 1 [en] (SIRT1) estimula a autofagia ao impedir a acetilação de proteínas (por meio da desacetilação) necessárias para a autofagia, conforme demonstrado em células cultivadas e tecidos embrionários e neonatais.[32] Essa função estabelece uma ligação entre a expressão da sirtuína e a resposta celular à limitação de nutrientes devido à restrição calórica.[33]
Outras funções biológicas
Além de participar do processo natural de reciclagem de estruturas, em outras determinadas condições fisiológicas, pode ocorrer um aumento da autofagia. Isso acontece, por exemplo, nas glândulas mamárias quando termina a lactação. Durante a gravidez, aumenta o número de células secretoras dessas glândulas, para produzir leite após o parto. Terminado o período de lactação, ocorre a destruição autofágica dos restos de secreção e das organelas não mais necessárias.
As enzimas lisossômicas, algumas vezes, também participam da digestão de moléculas extracelulares. Um aumento nos níveis citosólicos de Ca2+ induz a fusão da membrana dos lisossomos com a membrana plasmática, fazendo com que as enzimas sejam exocitadas no meio extracelular. Essa secreção das enzimas lisossômicas ocorre em condições normais, como, por exemplo, na remodelação dos ossos durante o crescimento, quando as enzimas lisossômicas digerem a matriz óssea para possibilitar o crescimento do esqueleto.
Células envolvidas em defesa e resposta imune, tais como os basófilos, eosinófilos mastócitos e linfócitos, apresentam compartimentos lisossômicos especializados, denominados lisossomos secretores. Esses lisossomos realizam a secreção regulada das suas enzimas, ou seja, secretam apenas em resposta a um estímulo externo. Eles desempenham duas funções: estocam as enzimas lisossômicas que, no momento apropriado, são secretadas no meio extracelular e inserem na membrana plasmática moléculas envolvidas no processo da resposta imune.
A autofagia degrada organelas, membranas celulares e proteínas danificadas, e acredita-se que a autofagia insuficiente seja uma das principais razões para o acúmulo de células danificadas e o envelhecimento.[34] A autofagia e os reguladores da autofagia estão envolvidos na resposta a danos lisossomais, frequentemente direcionados por galectinas [en], como a galectina-3 [en] e a galectina-8.[34]
Ver também
Referências
- 1 2 3 Levine B, Kroemer G (2019). «Biological Functions of Autophagy Genes: A Disease Perspective». Cell. 176 (1–2): 11–42. PMC 6347410
 . PMID 30633901. doi:10.1016/j.cell.2018.09.048
. PMID 30633901. doi:10.1016/j.cell.2018.09.048 - ↑ Kuo C, Hansen M, Troemel E (2018). «Autophagy and innate immunity: Insights from invertebrate model organisms». Autophagy. 14 (2): 33–242. PMC 5902216. PMID 29130360. doi:10.1080/15548627.2017.1389824
- ↑ Metur SP, Lei Y, Zhang Z, Klionsky DJ (2023). «Regulation of autophagy gene expression and its implications in cancer». Journal of Cell Science. 136 (10). PMC 10214848. PMID 37199330. doi:10.1242/jcs.260631
- ↑ Klionsky DJ (setembro de 2012). «Look people, "Atg" is an abbreviation for "autophagy-related." That's it». Autophagy. 8 (9): 1281–2. PMC 3442874. PMID 22889836. doi:10.4161/auto.21812
- ↑ Klionsky DJ, Cueva R, Yaver DS (outubro de 1992). «Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway». The Journal of Cell Biology. 119 (2): 287–99. PMC 2289658. PMID 1400574. doi:10.1083/jcb.119.2.287
- ↑ Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y (outubro de 1992). «Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction». The Journal of Cell Biology. 119 (2): 301–11. PMC 2289660. PMID 1400575. doi:10.1083/jcb.119.2.301
- ↑ Thumm M, Egner R, Koch B, Schlumpberger M, Straub M, Veenhuis M, Wolf DH (agosto de 1994). «Isolation of autophagocytosis mutants of Saccharomyces cerevisiae». FEBS Letters. 349 (2): 275–80. Bibcode:1994FEBSL.349..275T. PMID 8050581. doi:10.1016/0014-5793(94)00672-5
- ↑ Tsukada M, Ohsumi Y (outubro de 1993). «Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae». FEBS Letters. 333 (1–2): 169–74. Bibcode:1993FEBSL.333..169T. PMID 8224160. doi:10.1016/0014-5793(93)80398-e
- ↑ Harding TM, Morano KA, Scott SV, Klionsky DJ (novembro de 1995). «Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway». The Journal of Cell Biology. 131 (3): 591–602. PMC 2120622. PMID 7593182. doi:10.1083/jcb.131.3.591
- ↑ Mizushima N, Yoshimori T, Ohsumi Y (10 de novembro de 2011). «The role of Atg proteins in autophagosome formation». Annual Review of Cell and Developmental Biology. 27 (1): 107–32. PMID 21801009. doi:10.1146/annurev-cellbio-092910-154005
- 1 2 Lamb CA, Yoshimori T, Tooze SA (dezembro de 2013). «The autophagosome: origins unknown, biogenesis complex». Nature Reviews. Molecular Cell Biology. 14 (12): 759–74. Bibcode:2013NRMCB..14..759L. PMID 24201109. doi:10.1038/nrm3696
- ↑ Suzuki H, Osawa T, Fujioka Y, Noda NN (abril de 2017). «Structural biology of the core autophagy machinery». Current Opinion in Structural Biology. 43: 10–17. PMID 27723509. doi:10.1016/j.sbi.2016.09.010
- ↑ Russell RC, Yuan HX, Guan KL (janeiro de 2014). «Autophagy regulation by nutrient signaling». Cell Research. 24 (1): 42–57. PMC 3879708. PMID 24343578. doi:10.1038/cr.2013.166
- ↑ Chan EY (setembro de 2012). «Regulation and function of uncoordinated-51 like kinase proteins». Antioxidants & Redox Signaling. 17 (5): 775–85. PMID 22074133. doi:10.1089/ars.2011.4396
- ↑ Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (julho de 2013). «ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase». Nature Cell Biology. 15 (7): 741–50. Bibcode:2013NaCB...15..741R. PMC 3885611. PMID 23685627. doi:10.1038/ncb2757
- ↑ Itakura E, Kishi C, Inoue K, Mizushima N (dezembro de 2008). «Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG». Molecular Biology of the Cell. 19 (12): 5360–72. PMC 2592660. PMID 18843052. doi:10.1091/mbc.E08-01-0080
- ↑ Kang R, Zeh HJ, Lotze MT, Tang D (abril de 2011). «The Beclin 1 network regulates autophagy and apoptosis». Cell Death and Differentiation. 18 (4): 571–80. PMC 3131912. PMID 21311563. doi:10.1038/cdd.2010.191
- ↑ Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, Pagliarini V, Matteoni S, Fuoco C, Giunta L, D'Amelio M, Nardacci R, Romagnoli A, Piacentini M, Cecconi F, Fimia GM (outubro de 2010). «The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy». The Journal of Cell Biology. 191 (1): 155–68. PMC 2953445. PMID 20921139. doi:10.1083/jcb.201002100
- ↑ Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N (maio de 2008). «FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells». The Journal of Cell Biology. 181 (3): 497–510. PMC 2364687. PMID 18443221. doi:10.1083/jcb.200712064
- ↑ Proikas-Cezanne T, Takacs Z, Dönnes P, Kohlbacher O (janeiro de 2015). «WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome». J Cell Sci. 128 (2): 207–17. PMID 25568150. doi:10.1242/jcs.146258 (inativo 4 de Dezembro de 2025)
- 1 2 Kumar, Suresh; Javed, Ruheena; Mudd, Michal; Pallikkuth, Sandeep; Lidke, Keith A.; Jain, Ashish; Tangavelou, Karthikeyan; Gudmundsson, Sigurdur Runar; Ye, Chunyan; Rusten, Tor Erik; Anonsen, Jan Haug (novembro de 2021). «Mammalian hybrid pre-autophagosomal structure HyPAS generates autophagosomes». Cell. 184 (24): 5950–69. PMC 8616855. PMID 34741801. doi:10.1016/j.cell.2021.10.017
- ↑ Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA (julho de 2014). «WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1». Molecular Cell. 55 (2): 238–52. PMC 4104028. PMID 24954904. doi:10.1016/j.molcel.2014.05.021
- ↑ Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y (dezembro de 2007). «The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy». The Journal of Biological Chemistry. 282 (52): 37298–302. Bibcode:2007JBiCh.28237298H. PMID 17986448. doi:10.1074/jbc.C700195200
- ↑ Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T (junho de 2004). «LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation». Journal of Cell Science. 117 (Pt 13): 2805–12. PMID 15169837. doi:10.1242/jcs.01131
- ↑ Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, Yoshimori T (novembro de 2008). «An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure». Molecular Biology of the Cell. 19 (11): 4651–9. PMC 2575160. PMID 18768752. doi:10.1091/mbc.e08-03-0312
- ↑ Park S, Choi SG, Yoo SM, Son JH, Jung YK (2014). «Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy». Autophagy. 10 (11): 1906–20. PMC 4502719. PMID 25483962. doi:10.4161/auto.32177
- ↑ Fader CM, Sánchez DG, Mestre MB, Colombo MI (dezembro de 2009). «TI-VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways». Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1793 (12): 1901–16. PMID 19781582. doi:10.1016/j.bbamcr.2009.09.011
- ↑ Furuta N, Fujita N, Noda T, Yoshimori T, Amano A (março de 2010). «Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes». Molecular Biology of the Cell. 21 (6): 1001–10. PMC 2836953. PMID 20089838. doi:10.1091/mbc.e09-08-0693
- ↑ Kim YM, Jung CH, Seo M, Kim EK, Park JM, Bae SS, Kim DH (janeiro de 2015). «mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation». Molecular Cell. 57 (2): 207–18. PMC 4304967. PMID 25533187. doi:10.1016/j.molcel.2014.11.013
- ↑ Satoo K, Noda NN, Kumeta H, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F (maio de 2009). «The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy». The EMBO Journal. 28 (9): 1341–50. PMC 2683054. PMID 19322194. doi:10.1038/emboj.2009.80
- ↑ Yang Z, Huang J, Geng J, Nair U, Klionsky DJ (dezembro de 2006). «Atg22 recycles amino acids to link the degradative and recycling functions of autophagy». Molecular Biology of the Cell. 17 (12): 5094–104. PMC 1679675. PMID 17021250. doi:10.1091/mbc.e06-06-0479
- ↑ Yessenkyzy A, Saliev T, Zhanaliyeva M, Nurgozhin T (2020). «Polyphenols as Caloric-Restriction Mimetics and Autophagy Inducers in Aging Research». Nutrients. 12 (5): 1344. PMC 7285205. PMID 32397145. doi:10.3390/nu12051344
- ↑ Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T (março de 2008). «A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy». Proceedings of the National Academy of Sciences of the United States of America. 105 (9): 3374–89. Bibcode:2008PNAS..105.3374L. PMC 2265142. PMID 18296641. doi:10.1073/pnas.0712145105
- 1 2 Cuervo AM, Bergamini E, Brunk UT, Dröge W, Ffrench M, Terman A (2005). «Autophagy and aging: the importance of maintaining "clean" cells». Autophagy. 1 (3): 131–40. PMID 16874025. doi:10.4161/auto.1.3.2017
Bibliografia
- Cooper, Geoffrey M.; Hausman, Robert E. (2016). A Célula – Uma Abordagem Molecular 3 ed. Porto Alegre, Brasil: Artmed Editora. ISBN 0-87893-214-3
Ligações externas
| Na medicina |
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Listas |
| ||||||||||||
| Mortalidade |
| ||||||||||||
| Após a morte |
| ||||||||||||
| Paranormal | |||||||||||||
| Legal |
| ||||||||||||
| Áreas |
| ||||||||||||
| Outros |
| ||||||||||||
| |||||||||||||