UPGMA
UPGMA (unweighted pair group method with arithmetic mean) (em português: método de grupo de pares não ponderados com média aritmética) é um método de agrupamento hierárquico aglomerativo (de baixo para cima) simples. Existe também uma variante ponderada, WPGMA, e os dois métodos são geralmente atribuídos a Sokal e Michener.[1]
Observe-se que ser não ponderado indica que todas as distâncias contribuem igualmente para cada média calculada e não se referindo portanto, à matemática através da qual é obtida. Assim, a média simples no WPGMA produz um resultado ponderado e a média proporcional no UPGMA produz um resultado não ponderado ( veja o exemplo prático ).[2]
Algoritmo
O algoritmo UPGMA constrói uma árvore com raiz ( dendrograma ) que reflete a estrutura presente numa matriz de similaridade de pares (ou uma matriz de dissimilaridade ). Em cada etapa, os dois clusters mais próximos são combinados em um cluster de nível superior. A distância entre quaisquer dois clusters e , cada um de tamanho ( ou seja, cardinalidade ) e , é considerada a média de todas as distâncias entre pares de objetos em e em , ou seja, a distância média entre os elementos de cada cluster:








Por outras palavras, em cada etapa de agrupamento, a distância atualizada entre os clusters unidos e um novo cluster é dada pela média proporcional das distâncias e :





O algoritmo UPGMA produz dendrogramas com raiz e assume uma taxa constante, ou seja, assume uma árvore ultramétrica na qual as distâncias da raiz até ao fim de cada ramo são iguais. Quando as pontas são dados moleculares ( ou seja, DNA, RNA e proteína ) amostrados ao mesmo tempo, a suposição de ultrametricidade torna-se equivalente a assumir um relógio molecular.
Exemplo prático
Este exemplo prático é baseado numa matriz de distâncias genética JC69 calculada a partir do alinhamento da sequência de RNA ribossômico 5S de cinco bactérias: Bacillus subtilis ( ), Bacillus stearothermophilus ( ), Lactobacillus viridescens ( ), Acholeplasma modicum ( ) e Micrococcus luteus ( ).[3][4]





Primeiro passo
- Primeiro agrupamento
Assumindo que temos cinco elementos e a seguinte matriz de distâncias em pares entre eles:


| a | b | c | e | e | |
|---|---|---|---|---|---|
| a | 0 | 17 | 21 | 31 | 23 |
| b | 17 | 0 | 30 | 34 | 21 |
| c | 21 | 30 | 0 | 28 | 39 |
| e | 31 | 34 | 28 | 0 | 43 |
| e | 23 | 21 | 39 | 43 | 0 |
Neste exemplo, é o menor valor de , então juntamos os elementos e .

- Estimativa do comprimento do primeiro ramo
De ressaltar que denota o nó ao qual e estão agora conectados. Com é garantido que os elementos e são equidistantes de . Isto corresponde à expectativa da hipótese de ultrametricidade. Os ramos que unem e a têm então comprimentos ( veja o dendrograma final )



- Primeira atualização da matriz de distâncias
Em seguida, procedemos à atualização da matriz de distâncias inicial para uma nova matriz de distância (veja abaixo), reduzida em tamanho por uma linha e uma coluna devido ao agrupamento de com . Valores em negrito em correspondem às novas distâncias, calculadas pela média das distâncias entre cada elemento do primeiro par e cada um dos elementos restantes:





Valores em itálico em não são afetados pela atualização da matriz, já que correspondem a distâncias entre elementos não envolvidos no primeiro cluster.
Segundo passo
- Segundo agrupamento
Os três passos anteriores são então repetidos, começando agora com a nova matriz de distâncias
| (a,b) | c | e | e | |
|---|---|---|---|---|
| (a,b) | 0 | 25,5 | 32,5 | 22 |
| c | 25,5 | 0 | 28 | 39 |
| e | 32,5 | 28 | 0 | 43 |
| e | 22 | 39 | 43 | 0 |
Aqui, é o menor valor de , então juntamos o cluster e o elemento .

- Estimativa do comprimento do segundo ramo
Com a denotar o nó pelo qual e estão agora conectados. Devido à restrição de ultrametricidade, os ramos que unem ou a , e a são iguais e têm o seguinte comprimento:


Deduzimos o comprimento do ramo restante: ( veja o dendrograma final )

- Atualização da matriz de segunda distância
Em seguida, procedemos à atualização em uma nova matriz de distância (veja abaixo), reduzida em tamanho por uma linha e uma coluna devido ao agrupamento de com . Valores em negrito em correspondem às novas distâncias, calculadas pela média proporcional:


Graças a esta média proporcional, o cálculo desta nova distância tem em conta o maior tamanho do cluster (dois elementos) em relação a (um elemento). Da mesma forma:

A média proporcional, portanto, dá peso igual às distâncias iniciais da matriz . Esta é a razão pela qual o método não é ponderado, não devido ao procedimento matemático, mas em relação às distâncias iniciais.
Terceiro passo
- Terceiro agrupamento
Reiteramos novamente os três passos anteriores, partindo da matriz de distância atualizada .
| ((a,b),e) | c | e | |
|---|---|---|---|
| ((a,b),e) | 0 | 30 | 36 |
| c | 30 | 0 | 28 |
| e | 36 | 28 | 0 |
Aqui, é o menor valor de , então juntamos os elementos e .

- Estimativa do comprimento do terceiro ramo
Com a denotar o nó ao qual e estão agora conectados, os ramos que unem e a têm então comprimentos ( veja o dendrograma final )


- Atualização da matriz de terceira distância
Apenas uma entrada precisa de ser atualizada, considerando que os dois elementos e contribuem cada um com para o cálculo da média:


Etapa final
A final matriz é:

| ((a,b),e) | (cd) | |
|---|---|---|
| ((a,b),e) | 0 | 33 |
| (cd) | 33 | 0 |
Então juntamos os grupos e .


denota o nó (raiz) ao qual e estão agora conectados. Os ramos que unem e a têm então comprimentos:


Deduzimos os dois comprimentos de ramificação restantes:


O dendrograma UPGMA
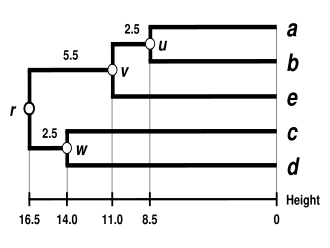
O dendrograma está agora completo.[5] É ultramétrico porque todas as pontas ( a ) são equidistantes de :

O dendrograma tem portanto como raiz, o seu nó mais basal.
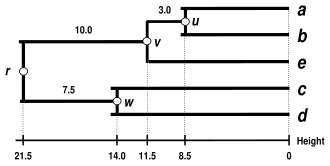
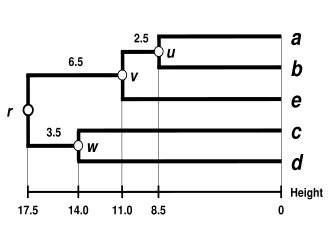
Comparação com outras ligações
Esquemas de ligação alternativos incluem agrupamento de ligação simples, agrupamento de ligação completa e agrupamento de ligação média WPGMA . Implementar uma ligação diferente é simplesmente uma questão de usar uma fórmula diferente para calcular distâncias entre clusters durante as etapas de atualização da matriz de distâncias do algoritmo acima. O agrupamento de ligação completa evita uma desvantagem do método alternativo de agrupamento de ligação única - o chamado fenômeno de encadeamento, onde os agrupamentos formados por meio do agrupamento de ligação única podem ser forçados a unir-se devido a elementos individuais estarem próximos uns dos outros, mesmo que muitos dos elementos em cada agrupamento possam estar muito distantes uns dos outros. A ligação completa tende a encontrar aglomerados compactos de diâmetros aproximadamente iguais.[6]
 |
|
 |
|
| Single-linkage clustering | Complete-linkage clustering | Average linkage clustering: WPGMA | Average linkage clustering: UPGMA. |
Usos
- Em ecologia, é um dos métodos mais populares para a classificação de unidades de amostragem (como parcelas de vegetação) com base na sua semelhança em variáveis descritivas relevantes (como a sua composição de espécies).[7] Por exemplo, tem sido usado para compreender a interação trófica entre bactérias marinhas e protistas.[8]
- Em bioinformática, o UPGMA é usado na criação de árvores fenéticas (fenogramas). O UPGMA foi inicialmente projetado para uso em estudos de eletroforese de proteínas, mas atualmente é mais frequentemente usado para produzir árvores-guia para algoritmos mais sofisticados. Este algoritmo é usado, por exemplo, em procedimentos de alinhamento de sequências, pois propõe uma ordem na qual as sequências serão alinhadas. Na verdade, a árvore guia visa agrupar as sequências mais semelhantes, independentemente da sua taxa evolutiva ou afinidades filogenéticas, e é exatamente esse o objetivo do UPGMA[9]
- Em filogenética, o UPGMA assume uma taxa constante de evolução ( hipótese do relógio molecular ) e que todas as sequências foram amostradas ao mesmo tempo, e não é um método bem conceituado para inferir relacionamentos, a menos que essa suposição tenha sido testada e justificada para o conjunto de dados usado. Observe que, mesmo sob um "relógio estrito", sequências amostradas em momentos diferentes não devem levar a uma árvore ultramétrica.
Complexidade temporal
Uma implementação trivial do algoritmo para construir a árvore UPGMA tem complexidade temporal e usar um heap para cada cluster para manter suas distâncias de outro cluster reduz seu tempo para . Fionn Murtagh apresentou um algoritmo de tempo e espaço.[10]



Veja também
- Método de Neighbor joining
- Análise de clusters
- Agrupamento de ligação única
- Agrupamento de ligação completa
- Agrupamento hierárquico
- Modelos de evolução do DNA
- Relógio molecular
Referências
- ↑ Sokal, Michener (1958). «A statistical method for evaluating systematic relationships». University of Kansas Science Bulletin. 38: 1409–1438 Erro no estilo Vancouver: wikilink (ajuda)
- ↑ Garcia, Santi; Puigbò, Pere. «DendroUPGMA: A dendrogram construction utility» (PDF). p. 4
- ↑ Erdmann VA, Wolters J (1986). «Collection of published 5S, 5.8S and 4.5S ribosomal RNA sequences». Nucleic Acids Research. 14 Suppl (Suppl): r1–59. PMC 341310
 . PMID 2422630. doi:10.1093/nar/14.suppl.r1
. PMID 2422630. doi:10.1093/nar/14.suppl.r1
- ↑ «Phylogenetic analysis using ribosomal RNA». Ribosomes. Col: Methods in Enzymology. 164. [S.l.: s.n.] 1988. pp. 793–812. ISBN 978-0-12-182065-7. PMID 3241556. doi:10.1016/s0076-6879(88)64084-5
- ↑ Swofford, David L.; Olsen, Gary J.; Waddell, Peter J.; Hillis, David M. (1996). «Phylogenetic inference». In: Hillis; Moritz, Craig; Mable, Barbara K. Molecular Systematics, 2nd edition. Sunderland, MA: Sinauer. pp. 407–514. ISBN 9780878932825
- ↑ Everitt, B. S.; Landau, S.; Leese, M. (2001). Cluster Analysis. 4th Edition. London: Arnold. pp. 62–64
- ↑ Numerical Ecology. Col: Developments in Environmental Modelling. 20 Second English ed. Amsterdam: Elsevier. 1998
- ↑ Vázquez-Domínguez E, Casamayor EO, Català P, Lebaron P (abril de 2005). «Different marine heterotrophic nanoflagellates affect differentially the composition of enriched bacterial communities». Microbial Ecology. 49 (3): 474–85. Bibcode:2005MicEc..49..474V. JSTOR 25153200. PMID 16003474. doi:10.1007/s00248-004-0035-5
- ↑ Wheeler TJ, Kececioglu JD (julho de 2007). «Multiple alignment by aligning alignments». Bioinformatics. 23 (13): i559–68. PMID 17646343. doi:10.1093/bioinformatics/btm226
- ↑ Murtagh F (1984). «Complexities of Hierarchic Clustering Algorithms: the state of the art». Computational Statistics Quarterly. 1: 101–113