Síndrome do QT longo
| Síndrome do QT longo | |
|---|---|
 | |
| Especialidade | cardiologia |
| Classificação e recursos externos | |
| CID-11 | BC65.0 |
| CID-10 | I45.81 |
| CID-9 | 426.82 |
| OMIM | 192500 |
| DiseasesDB | 11104 |
| eMedicine | 157826 |
| MeSH | D008133 |
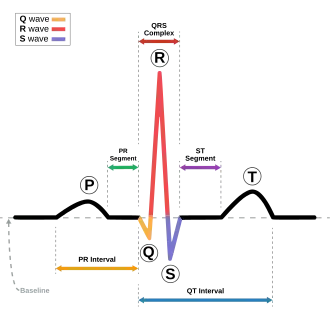
Síndrome do QT longo é um tipo de taquiarritimia ventricular congênita. Caracteriza-se por um alongamento do espaço QT (decorrente de alterações nos canais iônicos miocárdico) no eletrocardiograma de surfície, evento associado a um risco elevado de torsades de pointes ou de fibrilação ventricular podendo evoluir para uma síncope, flutter, PCR e morte súbita.[1]
Pode se apresentar de maneira isolada ou associada a várias malformações, como na síndrome de Romano-Ward, mais comum, ou na síndrome de Jervell e Lange-Nielsen, menos frequente, ligada à surdez, nos casos autossómicos recessivos. Pode ainda ocorrer de forma adquirida, provocada por medicamentos antiarrítmicos do grupos IA, IB e III ou por alguns anti-histamínicos.
Geralmente é diagnosticada após um evento cardíaco subsequente a exercício físico ou crise emocional, por exemplo.
Epidemiologia e Incidência
A prevalência da síndrome do QT longo é estimada em aproximadamente 1 para cada 2.000 nascidos vivos, embora existam muitos casos de subdiagnóstico devido a portadores assintomáticos. A condição não apresenta predileção clara por etnia, mas observa-se uma frequência discretamente maior em mulheres. É considerada uma das principais causas de morte súbita cardíaca em crianças e adultos jovens com corações estruturalmente normais, sendo um ponto de atenção crucial em exames de rotina e medicina esportiva.
Fisiopatologia e Mecanismos Celulares
O prolongamento do intervalo QT reflete um atraso na repolarização ventricular, causado por disfunções proteicas nos canais iônicos da membrana celular (canalopatias). Essas alterações podem ser de "perda de função" (geralmente nos canais de potássio) ou "ganho de função" (nos canais de sódio). Esse atraso cria um estado de instabilidade elétrica que favorece o surgimento de pós-despolarizações precoces, que funcionam como gatilhos para arritmias ventriculares polimórficas graves, como a Torsades de Pointes.
Classificação Genética e Subtipos
A SQTL congênita é dividida em diversos subtipos, baseados no gene específico que sofreu a mutação.
Subtipos Principais (LQT1, LQT2 e LQT3): Representam cerca de 90% dos casos e focam essencialmente em arritmias cardíacas desencadeadas por exercício físico (LQT1), sustos auditivos (LQT2) ou durante o sono e repouso (LQT3).
Subtipos Raros e suas Complicações (LQT4 a LQT15): Estes subtipos são menos frequentes, mas costumam apresentar complicações que afetam outros sistemas do organismo:
SQTL4 (Gene ANK2): Pode causar disfunção severa no nó sinusal, bradicardia extrema e fibrilação atrial.
SQTL7 (Síndrome de Andersen-Tawil): Apresenta uma tríade de complicações: QT longo, paralisia periódica muscular e malformações físicas (mandíbula pequena, orelhas baixas e dedos curvados).
SQTL8 (Síndrome de Timothy): Uma das formas mais graves, com complicações como malformações cardíacas estruturais, dedos fundidos (sindactilia), deficiência imunológica e distúrbios do espectro autista.
SQTL10 a LQT15: Envolvem mutações em proteínas acessórias e geralmente apresentam maior resistência aos tratamentos convencionais, exigindo terapias combinadas.
Critérios de Diagnóstico
O diagnóstico baseia-se na combinação de achados eletrocardiográficos, histórico clínico e antecedentes familiares, frequentemente sistematizados pelo Escore de Schwartz.
Eletrocardiograma: O Intervalo QT corrigido (QTc) é calculado pela fórmula de Bazett. Valores de QTc acima de 470ms em homens e 480ms em mulheres são indicadores diagnósticos fortes.
Histórico Familiar: Presença de parentes de primeiro grau com SQTL confirmada ou casos de morte súbita inexplicada antes dos 30 anos de idade.
Prognóstico
É fundamental compreender que a Síndrome do QT Longo não tem cura. Por ser uma condição genética que altera permanentemente as proteínas dos canais iônicos, ela acompanha o indivíduo por toda a vida. No entanto, o prognóstico é excelente com o manejo adequado, cujo objetivo não é eliminar a mutação, mas prevenir complicações fatais. As crises podem ser evitadas através de:
Farmacoterapia: Uso contínuo de betabloqueadores para estabilizar o ritmo cardíaco.
Estilo de Vida: Restrição absoluta de medicamentos que prolongam o QT e monitoramento de eletrólitos (potássio e magnésio).
Intervenção: Implante de Cardioversor Desfibrilador (CDI) para pacientes de alto risco.
Aconselhamento Genético
Devido ao caráter hereditário, geralmente autossômico dominante, o diagnóstico de um paciente exige a investigação de seus familiares próximos. Isso permite que portadores "silenciosos" sejam identificados e tratados preventivamente.
Fonte Consultada:
Referências
| Controle de autoridade |
|---|