Pegada de massas peptídicas
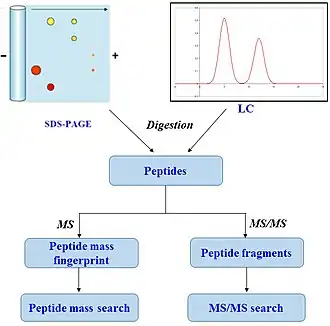
A pegada de massa de peptídeos (PMF), também chamada de pegada de peptídeos ou impressão digital da massa peptídica, é uma técnica analítica para a identificação de proteínass na qual a proteína desconhecida de interesse é primeiro clivada em pequenos peptídeos, cujas massas absolutas são medidas com precisão com um espectrómetro de massa como MALDI-TOF ou ESI-TOF.[1] O método foi desenvolvido em 1993 por vários grupos de forma independente.[2][3][4][5][6] As massas dos peptídeos são comparadas a uma base de dados de sequências de proteínas conhecidas ou mesmo ao genoma. Isto é feito através de programas de computador que traduzem o genoma conhecido do organismo em proteínas, depois cortam teoricamente as proteínas em peptídeos e calculam as massas absolutas dos peptídeos de cada proteína. As massas dos peptídeos das proteínas desconhecidas são então comparadas com as massas teóricas dos peptídeos de cada proteína codificada no genoma. Os resultados são analisados estatisticamente para encontrar a melhor correspondência.
A vantagem deste método é que apenas as massas dos peptídeos precisam de ser conhecidas. Uma desvantagem é que a sequência proteica deve estar presente na base de dados de interesse. Além disso, a maioria dos algoritmos para a identificação de peptídeos através da análise de massas pressupõe que os peptídeos provêm de uma única proteína.[7] A presença de uma mistura pode complicar significativamente a análise e comprometer os resultados. Um requisito típico para a identificação de proteínas com base na análise de pegada de massa de peptídeos é a obtenção de uma proteína isolada. As misturas com mais de 2 a 3 proteínas requerem frequentemente o uso adicional de espectrometria de massas em tandem (MS/MS) para alcançar especificidade suficiente. Assim sendo, as amostras típicas para a análise de pegada de massa de peptídeos são proteínas isoladas por eletroforese bidimensional em gel (géis 2D) ou bandas isoladas por SDS-PAGE. Análises adicionais de MS/MS podem ser diretas, como por exemplo, análises MALDI-TOF/TOF, ou análises subsequentes por nanoLC-ESI-MS/MS dos eluatos das manchas de gel.[7][8]
Origens
A análise de pegadas de massas de peptídeos foi desenvolvida devido ao longo e tedioso processo de análise de proteínas. A degradação de Edman, utilizada na análise de proteínas, demorava quase uma hora a analisar um resíduo de aminoácido.[9] A SDS-PAGE também tem sido utilizada para separar proteínas em misturas muito complexas, que também empregam electrotransferência e métodos de coloração.[10] As bandas são então extraídas do gel e sequenciadas automaticamente. Um problema recorrente no processo era que as proteínas interferentes eram também purificadas juntamente com a proteína de interesse. As sequências destas proteínas interferentes foram compiladas no que ficou conhecido como base de dados Dayhoff.[11] Por fim, a presença das sequências destas proteínas contaminantes nas bases de dados reduziu o tempo instrumental e o custo da análise proteica.
Preparação da amostra
As amostras de proteínas podem ser obtidas por SDS-PAGE[7] ou HPLC de fase reversa e depois submetidas a algumas modificações químicas. As ligações dissulfeto das proteínas são reduzidas e os aminoácidos cisteína são quimicamente carbamidometilados ou acrilamidizados durante a eletroforese em gel.
As proteínas são depois clivadas em vários fragmentos utilizando enzimas proteolíticas (proteasess) como a tripsina, quimotripsina ou Glu-C. Uma relação amostra:protease típica é de 50:1. A proteólise é geralmente realizada durante a noite e os peptídeos resultantes são extraídos com acetonitrila e secos a vácuo. Os péptidos são depois dissolvidos numa pequena quantidade de água destilada ou ainda mais concentrados e purificados, estando prontos para análise por espectrometria de massas.
Análise por espectrometria de massas
A proteína digerida pode ser analisada com diferentes tipos de espectrómetros de massas, como o ESI-TOF ou o MALDI-TOF. O MALDI-TOF é geralmente o instrumento preferido, pois permite uma elevada produtividade das amostras e a análise de múltiplas proteínas numa única experiência, se complementado com análise MS/MS. A LC/ESI-MS e a CE/ESI-MS são também excelentes técnicas para a identificação de péptidos por espectrometria de massas.[12][13]
Uma pequena fracção do peptídeo (geralmente 1 microlitro ou menos) é pipetada sobre uma placa alvo MALDI e um composto químico denominado matriz MALDI é adicionado à mistura de peptídeos. As matrizes comuns são ácido sinapínico, ácido alfa-ciano-4-hidroxicinâmico e ácido 2,3-di-hidroxibenzoico. As moléculas da matriz são necessárias para a dessorção das moléculas de peptídeo. A matriz e as moléculas de peptídeo co-cristalizam na placa alvo MALDI e estão prontas para análise. Existe uma técnica predominante de preparação de amostras para MALDI-MS denominada técnica da gota seca.[14] O alvo é inserido na câmara de vácuo do espectrómetro de massas e a dessorção e ionização dos fragmentos peptídicos é iniciada por um feixe de laser pulsado que transfere grandes quantidades de energia para as moléculas da matriz. A transferência de energia é suficientemente grande para promover a ionização e a transição das moléculas da matriz e dos péptidos da fase sólida para a fase gasosa. Os iões são acelerados no campo elétrico do espectrómetro de massas e deslocam-se em direção a um detetor de iões, onde a sua chegada é detetada como um sinal elétrico. A relação massa/carga é proporcional ao seu tempo de voo (TOF) no tubo de deriva e pode ser calculada a partir deste.
Ao acoplar a ionização por electrospray (ESI) com a cromatografia líquida capilar (LC), os péptidos podem ser separados das proteínas digeridas, enquanto as suas massas moleculares são obtidas simultaneamente.[15] Outra técnica é a eletroforese capilar acoplada à espectrometria de massas com ionização por electrospray (ESI-MS); no entanto, funciona melhor na análise de pequenas quantidades de proteína.
Análise computacional
A análise por espectrometria de massas produz uma lista de pesos moleculares, frequentemente designada por lista de picos. As massas dos peptídeos são comparadas com bases de dados de proteínas, como o SwissProt, que contém informação sobre a sequência proteica. O software realiza simulações virtuais "in silico" da digestão das proteínas da base de dados com a mesma enzima (por exemplo, tripsina) utilizada na reação de clivagem química real. A massa destes peptídeos é então calculada e comparada com a lista de picos de massas medidas. Os resultados são analisados estatisticamente e as possíveis correspondências são apresentadas numa tabela de resultados.
Ver também
Referências
- ↑ Clauser KR, Baker P, Burlingame AL (1999). «Role of accurate mass measurement (+/- 10 ppm) in protein identification strategies employing MS or MS/MS and database searching». Anal. Chem. 71 (14): 2871–82. PMID 10424174. doi:10.1021/ac9810516
- ↑ Pappin DJ, Hojrup P, Bleasby AJ (1993). «Rapid identification of proteins by peptide-mass fingerprinting». Curr. Biol. 3 (6): 327–32. Bibcode:1993CBio....3..327P. PMID 15335725. doi:10.1016/0960-9822(93)90195-T
- ↑ Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, Watanabe C (1993). «Identifying proteins from two-dimensional gels by molecular mass searching of peptide fragments in protein sequence databases». Proc. Natl. Acad. Sci. U.S.A. 90 (11): 5011–5. Bibcode:1993PNAS...90.5011H. PMC 46643
 . PMID 8506346. doi:10.1073/pnas.90.11.5011
. PMID 8506346. doi:10.1073/pnas.90.11.5011
- ↑ Mann M, Højrup P, Roepstorff P (1993). «Use of mass spectrometric molecular weight information to identify proteins in sequence databases». Biological Mass Spectrometry. 22 (6): 338–45. PMID 8329463. doi:10.1002/bms.1200220605
- ↑ James P, Quadroni M, Carafoli E, Gonnet G (1993). «Protein identification by mass profile fingerprinting». Biochem. Biophys. Res. Commun. 195 (1): 58–64. PMID 8363627. doi:10.1006/bbrc.1993.2009
- ↑ Yates JR, Speicher S, Griffin PR, Hunkapiller T (1993). «Peptide mass maps: a highly informative approach to protein identification». Anal. Biochem. 214 (2): 397–408. PMID 8109726. doi:10.1006/abio.1993.1514
- ↑ a b c Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, Wilm M, Vorm O, Mortensen P, Shevchenko A, Boucherie H, Mann M (1996). «Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels». Proc. Natl. Acad. Sci. U.S.A. 93 (25): 14440–5. Bibcode:1996PNAS...9314440S. PMC 26151. PMID 8962070. doi:10.1073/pnas.93.25.14440
- ↑ Wang W, Sun J, Nimtz M, Deckwer WD, Zeng AP (2003). «Protein identification from two-dimensional gel electrophoresis analysis of Klebsiella pneumoniae by combined use of mass spectrometry data and raw genome sequences». Proteome Science. 1 (1). 6 páginas. PMC 317362. PMID 14653859. doi:10.1186/1477-5956-1-6
- ↑ Henzel, William J.; Watanabe, CA vantaxe deste método é queolin; Stults, John T. (1 de setembro de 2003). «Protein identification: The origins of peptide mass fingerprinting». Journal of the American Society for Mass Spectrometry (em inglês). 14 (9): 931–942. Bibcode:2003JASMS..14..931H. ISSN 1044-0305. PMID 12954162. doi:10.1016/S1044-0305(03)00214-9
- ↑ Matsudaira, P. (25 de julho de 1987). «Sequence from picomole quantities of proteins electroblotted onto polyvinylidene difluoride membranes». The Journal of Biological Chemistry. 262 (21): 10035–10038. ISSN 0021-9258. PMID 3611052. doi:10.1016/S0021-9258(18)61070-1
- ↑ B C Orcutt; D G George; Dayhoff, and M. O. (1983). «Protein and Nucleic Acid Sequence Database Systems». Annual Review of Biophysics and Bioengineering. 12 (1): 419–441. PMID 6347043. doi:10.1146/annurev.bb.12.060183.002223
- ↑ Moore, R. E.; Licklider, L.; Schumann, D.; Lee, T. D. (1 de dezembro de 1998). «A microscale electrospray interface incorporating a monolithic, poly(styrene-divinylbenzene) support for on-line liquid chromatography/tandem mass spectrometry analysis of peptides and proteins». Analytical Chemistry. 70 (23): 4879–4884. ISSN 0003-2700. PMID 9852776. doi:10.1021/ac980723p
- ↑ Whitmore, Colin D.; Gennaro, Lynn A. (1 de junho de 2012). «Capillary electrophoresis-mass spectrometry methods for tryptic peptide mapping of therapeutic antibodies». Electrophoresis (em inglês). 33 (11): 1550–1556. ISSN 1522-2683. PMID 22736356. doi:10.1002/elps.201200066
- ↑ Thiede, Bernd (2005). «Peptide mass fingerprinting». Methods. 35 (3): 237–247. PMID 15722220. doi:10.1016/j.ymeth.2004.08.015
- ↑ Dass, Chhabil (2007). Fundamentals of Contemporary Mass Spectrometry | Wiley Online Books (em inglês). [S.l.: s.n.] ISBN 9780470118498. doi:10.1002/0470118490