Homoquiralidade
Homoquiralidade é uma uniformidade de quiralidade, ou destreza. Objetos são quirais quando não podem ser superpostos em suas imagens espelhadas. Por exemplo, as mãos esquerda e direita de um humano são aproximadamente imagens espelhadas uma da outra, mas não são suas próprias imagens espelhadas, então são quirais. Em biologia, 19 dos 20 aminoácidos naturais são homoquirais, sendo L-quiral (canhotos), enquanto açúcares são D-quiral (destros).[1] Homoquiralidade pode também referir-se a substâncias enantiopuras em que todos os constituintes são os mesmos enantiômeros (uma versão destra ou canhota de um átomo ou molécula), mas algumas fontes desencorajam esse uso do termo.[2]
Não está claro se a homoquiralidade tem um propósito; no entanto, parece ser uma forma de armazenamento de informações.[3] Uma sugestão é que isso reduz as barreiras de entropia na formação de grandes moléculas organizadas.[4] Foi verificado experimentalmente que os aminoácidos formam grandes agregados em maior abundância a partir de amostras enantiopuras do aminoácido do que de amostras racêmicas (misturadas enantiomericamente).[4]
Não está claro se a homoquiralidade surgiu antes ou depois da vida, e muitos mecanismos para sua origem foram propostos.[5] Alguns desses modelos propõem três etapas distintas: quebra de simetria especular cria um pequeno desequilíbrio enantiomérico, amplificação quiral baseia-se neste desequilíbrio e transmissão quiral é a transferência de quiralidade de um conjunto de moléculas para outro.
Em biologia
Os aminoácidos são os blocos de construção de peptídeos e enzimas enquanto as cadeias de açúcar-peptídeo são a espinha dorsal de RNA e DNA.[6][7] Nos organismos biológicos, os aminoácidos aparecem quase exclusivamente na forma canhota (L-aminoácidos) e açúcares na forma destra (R-açúcares).[8] Como as enzimas catalisam reações, elas reforçam a homoquiralidade em uma grande variedade de outros produtos químicos, incluindo hormônios, toxinas, fragrâncias e sabores de alimentos.[9]:493–494 Glicina é aquiral, assim como alguns outros aminoácidos não-proteinogênico que são aquirais (tal como dimetilglicina) ou da forma enantiomérica D.
Organismos biológicos discriminam facilmente entre moléculas com diferentes quiralidades. Isso pode afetar reações fisiológicas como olfato e paladar. Carvona, um terpenóide encontrado nos óleos essenciais, tem cheiro de menta na forma L e alcaravia na forma R.[9]:494 Limoneno tem gosto de cítrico para destros e de pinho para canhotos.[10]:168
A homoquiralidade também afeta a resposta aos medicamentos. Talidomida, na sua forma canhota, cura enjoos matinais; na sua forma destra, causa defeitos de nascença.[10]:168 Infelizmente, mesmo que uma versão puramente canhota seja administrada, parte dela pode se converter para a forma destra no paciente.[11] Muitos medicamentos estão disponíveis como uma mistura racêmica (quantidades iguais de ambas as quiralidades) e uma medicamento enantiopuro (apenas uma quiralidade). Dependendo do processo de fabricação, as formas enantiopuras podem ser mais caras de produzir do que as misturas estereoquímicas.[10]:168
As preferências quirais também podem ser encontradas em um nível macroscópico. As conchas de caracóis podem ser hélices que viram para a direita ou para a esquerda, mas uma forma ou outra é fortemente preferida em uma determinada espécie. No caracol comestível Helix pomatia, somente um de 20 mil indivíduos é helicoidal à esquerda.[12]:61–62 O enrolamento das plantas pode ter uma quiralidade preferida e até mesmo o movimento de mastigação das vacas tem um excesso de 10% em uma direção.[13]
Origens
Quebra de simetria
As teorias para a origem da homoquiralidade nas moléculas da vida podem ser classificadas como determinísticas ou baseadas no acaso, dependendo do mecanismo proposto. Se existe uma relação entre causa e efeito — isto é, um campo quiral específico ou influência que causa a quebra da simetria especular — a teoria é classificada como determinística; caso contrário, é classificada como uma teoria baseada em mecanismos ao acaso (no sentido de aleatoriedade).[14]
Outra classificação para as diferentes teorias da origem da homoquiralidade biológica poderia ser feita dependendo se a vida surgiu antes da etapa de enantiodiscriminação (teorias bióticas) ou depois (teorias abióticas). As teorias bióticas afirmam que a homoquiralidade é simplesmente um resultado do processo natural de autoamplificação da vida—ou que a formação da vida preferindo uma quiralidade ou outra foi um evento raro e casual que aconteceu com as quiralidades que observamos, ou que todas as quiralidades da vida surgiram rapidamente, mas devido a eventos catastróficos e forte competição, as outras preferências quirais não observadas foram eliminadas pela preponderância e enriquecimento metabólico e enantiomérico das escolhas de quiralidade 'vencedoras'.[15][16] Se esse fosse o caso, restos do extinto sinal de quiralidade deveriam ser encontrados. Como esse não é o caso, hoje em dia as teorias bióticas não são mais apoiadas.
O surgimento do consenso de quiralidade como um processo natural de autoamplificação também foi associado à 2a lei da termodinâmica.[17]
Teorias determinísticas
Teorias determinísticas podem ser divididas em dois subgrupos: se a influência quiral inicial ocorreu em um espaço ou local de tempo específico (com média de zero em áreas de observação ou períodos de tempo suficientemente grandes), a teoria é classificada como determinística local; se a influência quiral for permanente no momento em que a seleção quiral ocorreu, então ela é classificada como determinística universal. Os grupos de classificação para teorias deterministas locais e teorias baseadas em mecanismos de acaso podem se sobrepor. Mesmo se uma influência quiral externa produzisse o desequilíbrio quiral inicial de forma determinística, o sinal do resultado poderia ser aleatório, já que a influência quiral externa tem sua contraparte enantiomérica em outro lugar.
Em teorias determinísticas, o desequilíbrio enantiomérico é criado devido a um campo ou influência quiral externa, e o sinal final impresso em biomoléculas será devido a ele. Mecanismos determinísticos para a produção de misturas não racêmicas a partir de materiais iniciais racêmicos incluem: leis físicas assimétricas, como a força eletrofraca (via raios cósmicos[18]) ou ambientes assimétricos, como os causados por luz polarizada circularmente, cristais de quartzo ou a rotação da Terra, β-Radiólise ou efeito magnetoquiral.[19][20] A teoria determinística universal mais aceita é a interação eletrofraca. Uma vez estabelecida, a quiralidade seria selecionada por ela.[21]
Uma suposição é que a descoberta de um desequilíbrio enantiomérico em moléculas no meteorito Murchison apoia uma origem extraterrestre da homoquiralidade: há evidências da existência de luz polarizada circularmente originando-se da dispersão de Mie em partículas de poeira interestelar alinhadas que podem desencadear a formação de uma excesso enantiomérico dentro do material quiral no espaço.[12]:123–124 Campos magnéticos interestelares e quase estelares podem alinhar partículas de poeira dessa maneira.[22] Outra especulação (a hipótese Vester-Ulbricht) sugere que a quiralidade fundamental dos processos físicos, como o decaimento beta (ver Violação de paridade) leva a meias-vidas ligeiramente diferentes de moléculas biologicamente relevantes.
Teorias do acaso
As teorias do acaso baseiam-se na suposição de que "Síntese assimétrica absoluta, i.e., a formação de produtos enantiomericamente enriquecidos a partir de precursores aquirais sem a intervenção de reagentes químicos quirais ou catalisadores é, na prática, inevitável apenas por motivos estatísticos".[23]
Considere o estado racêmico como uma propriedade macroscópica descrita por uma distribuição binomial; o experimento de lançar uma moeda, onde os dois resultados possíveis são os dois enantiômeros, é uma boa analogia. A distribuição de probabilidade discreta de obter n sucessos de Bernoulli trials, onde o resultado de cada ensaio de Bernoulli ocorre com probabilidade e o oposto ocorre com a probabilidade é dado por:




.

A distribuição de probabilidade discreta de ter exatamente moléculas de uma quiralidade e da outra, é dado por:


.

Como no experimento de lançar uma moeda, neste caso, assumimos ambos os eventos ( ou ) ser equiprovável, . A probabilidade de ter exatamente a mesma quantidade de ambos os enantiômeros é inversamente proporcional à raiz quadrada do número total de moléculas . Para um mol de um composto racêmico, moléculas, essa probabilidade se torna . A probabilidade de encontrar o estado racêmico é tão pequena que podemos considerá-la desprezível.





Neste cenário, há necessidade de amplificar o excesso enantiomérico estocástico inicial através de qualquer mecanismo eficiente de amplificação.[5] O caminho mais provável para esta etapa de amplificação é por autocatálise assimétrica. Uma reação química autocatalítica é aquela em que o produto da reação é ele próprio reativo, ou seja, uma reação química é autocatalítica se o produto da reação é ele próprio o catalisador da reação. Na autocatálise assimétrica, o catalisador é uma molécula quiral, o que significa que uma molécula quiral está catalisando sua própria produção. Um excesso enantiomérico inicial, como o que pode ser produzido pela luz polarizada, permite que o enantiômero mais abundante supere o outro.
Amplificação
Teoria
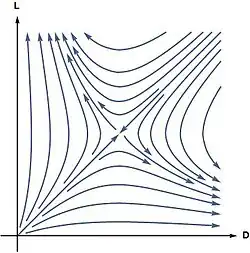
Em 1953, Charles Frank propôs um modelo para demonstrar que a homoquiralidade é uma consequência de autocatálise.[24][25] Em seu modelo os enantiômeros L e D de uma molécula quiral são produzidos autocataliticamente a partir de uma molécula aquiral A

enquanto se suprimiam mutuamente através de uma reação que ele chamou de antagonismo mútuo

Neste modelo, o estado racêmico é instável no sentido de que o menor excesso enantiomérico será amplificado para um estado completamente homoquiral. Isso pode ser demonstrado calculando as taxas de reação a partir da lei de ação das massas:
![{\displaystyle {\begin{aligned}{\frac {d[{\ce {L}}]}{dt}}&=k_{a}[{\ce {A}}][{\ce {L}}]-k_{d}{\ce {[L][D]}}\\{\frac {d[{\ce {D}}]}{dt}}&=k_{a}[{\ce {A}}][{\ce {D}}]-k_{d}{\ce {[L][D]}},\end{aligned}}}](./_assets_/eb734a37dd21ce173a46342d1cc64c92/2a8ae3fd5c4a5872bb86ef9127bd98381f5f9e12.svg)
onde é a constante de velocidade para as reações autocatalíticas, é a constante de velocidade para a reação de antagonismo mútuo e a concentração de A é mantida constante para simplificar.


As soluções analíticas para são encontradas como . A razão aumenta a uma taxa mais que exponencial se é positiva (e vice versa). Cada condição inicial é diferente
![{\displaystyle [L]/[D]=[L]_{0}/[D]_{0}\,e^{kd([L]_{0}-[D]_{0})(e^{k_{a}t}-1)}}](./_assets_/eb734a37dd21ce173a46342d1cc64c92/01844ec94b05c670cb4d46a4ec5956becb793c68.svg)
![{\displaystyle [L]/[D]}](./_assets_/eb734a37dd21ce173a46342d1cc64c92/439fcfce6c97c70ea345dfd7b0a0cee0ffa0d5e5.svg)
![{\displaystyle ([L]_{0}-[D]_{0})}](./_assets_/eb734a37dd21ce173a46342d1cc64c92/eceec5368fc7f05776d490c8869673a326de49c7.svg)
leva a uma das assíntotas ou . Assim a igualdade de e e assim por diante e representa uma condição de equilíbrio instável, sendo este resultado dependente da presença do termo que representa antagonismo mútuo.
![{\displaystyle [L]_{0}=[D]_{0}}](./_assets_/eb734a37dd21ce173a46342d1cc64c92/262f2ce1c587c2819c25d4f1a53c93a2ed8d08d0.svg)
![{\displaystyle [L]=0}](./_assets_/eb734a37dd21ce173a46342d1cc64c92/9432e9d1e600524a670e05b340c3bfe310cf12b8.svg)
![{\displaystyle [D]=0}](./_assets_/eb734a37dd21ce173a46342d1cc64c92/b1137d39625389c56402174e74b6a8de3da18864.svg)
![{\displaystyle [L]_{0}}](./_assets_/eb734a37dd21ce173a46342d1cc64c92/db821cb086b0448d2c4d4db2bc348d6a1e7ec81b.svg)
![{\displaystyle [D]_{0}}](./_assets_/eb734a37dd21ce173a46342d1cc64c92/9bb94024805d4f35f68e5b277483c654c315d934.svg)
![{\displaystyle [L]}](./_assets_/eb734a37dd21ce173a46342d1cc64c92/c1eba88cad4b17de726ed2c779b29eecdf5819a2.svg)
![{\displaystyle [D]}](./_assets_/eb734a37dd21ce173a46342d1cc64c92/1c4bcb25d77a0de74082c849178200b8cf1340b4.svg)
Ao definir o excesso enantiomérico como

![{\displaystyle ee={\frac {[{\ce {D}}]-[{\ce {L}}]}{[{\ce {D}}]+[{\ce {L}}]}},}](./_assets_/eb734a37dd21ce173a46342d1cc64c92/d7dee4e2fd16d609fd4442e804a26cf421c6ab27.svg)
a taxa de variação do excesso enantiomérico pode ser calculada usando regra da cadeia da taxa de variação das concentrações dos enantiômeros L e D.
![{\displaystyle {\frac {d(ee)}{dt}}=\left({\frac {2k_{d}{\ce {[L][D]}}}{[{\ce {D}}]+[{\ce {L}}]}}\right)ee.}](./_assets_/eb734a37dd21ce173a46342d1cc64c92/3471e4bec966fec2b85a90def8c59c45db0076fb.svg)
A análise de estabilidade linear desta equação mostra que o estado racêmico é instável. Começando em quase todos os lugares no espaço de concentração, o sistema evolui para um estado homoquiral.

É geralmente entendido que a autocatálise sozinha não produz homoquiralidade, e a presença da relação mutuamente antagônica entre os dois enantiômeros é necessária para a instabilidade da mistura racêmica. No entanto, estudos recentes mostram que a homoquiralidade pode ser alcançada a partir da autocatálise na ausência da relação mutuamente antagônica, mas o mecanismo subjacente para quebra de simetria é diferente.[5][26]
Experimentos
Existem vários experimentos de laboratório que demonstram como uma pequena quantidade de um enantiômero no início de uma reação pode levar a um grande excesso de um único enantiômero como produto. Por exemplo, a reação de Soai é autocatalítica.[27][28] Se a reação for iniciada com algum dos enantiômeros do produto já presente, o produto atua como um catalisador enantiosseletivo para produção de mais do mesmo enantiômero.[29] A presença inicial de apenas 0,2 equivalente de um enantiômero pode levar até 93% de um excesso enantiomérico do produto.
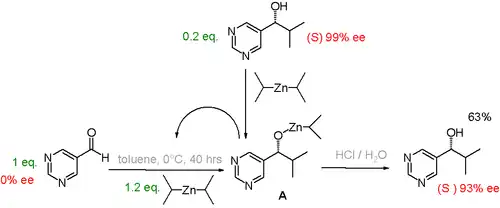
Outro estudo[30] diz respeito a aminoxilação catalisada por prolina do propionaldeído pelo nitrosobenzeno. Neste sistema, um pequeno excesso enantiomérico de catalisador leva a um grande excesso enantiomérico de produto.
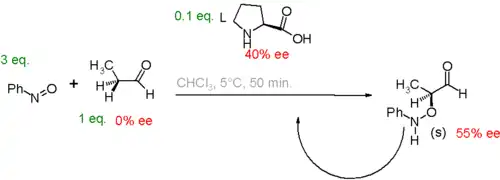
Clusters de octâmeros de serinas[31][32] também são concorrentes. Esses aglomerados de 8 moléculas de serina aparecem na espectrometria de massa com uma preferência homoquiral incomum; no entanto, não há evidências de que tais aglomerados existam em condições não ionizantes e o comportamento da fase de aminoácidos é muito mais relevante do ponto de vista prebiótico.[33] A observação recente de que sublimação parcial de uma amostra enantioenriquecida a 10% de leucina resulta em um enriquecimento de até 82% no sublimato mostra que enantioenriquecimento de aminoácidos pode ocorrer no espaço.[34] Processos de sublimação parcial podem ocorrer na superfície de meteoros onde existem grandes variações de temperatura. Esta descoberta pode ter consequências para o desenvolvimento do Mars Organic Detector do ExoMars programado para ser lançado em 2013, que visa recuperar quantidades vestigiais de aminoácidos da superfície de Marte exatamente por uma técnica de sublimação.
Uma alta amplificação assimétrica do excesso enantiomérico de açúcares também está presente na formação assimétrica de carboidratos catalisada por aminoácidos[35]
Um estudo clássico envolve um experimento que ocorre em laboratório.[36] Quando se permite que clorato de sódio cristalize em água e os cristais coletados examinados em um polarímetro, cada cristal acaba sendo quiral e a forma L ou a forma D. Em um experimento comum, a quantidade de cristais L coletados é igual à quantidade de cristais D (corrigido para efeitos estatísticos). Entretanto, quando a solução de clorato de sódio é agitada durante o processo de cristalização, os cristais são exclusivamente L ou exclusivamente D. Em 32 experimentos consecutivos de cristalização, 14 experimentos produziram cristais D e 18 outros cristais L. A explicação para essa quebra de simetria não é clara, mas está relacionada à autocatálise que ocorre no processo de nucleação.
Em um experimento relacionado, uma suspensão cristalina de um derivado aminoácido racêmico agitado continuamente, resulta em uma fase cristalina 100% de um dos enantiômeros porque o par enantiomérico é capaz de se equilibrar em solução (comparar com resolução cinética dinâmica).[37]
Transmissão
Uma vez que um enriquecimento enantiomérico significativo tenha sido produzido em um sistema, a transferência de quiralidade através de todo o sistema é costumeira. Esta última etapa é conhecida como etapa de transmissão quiral. Muitas estratégias em síntese assimétrica são construídas sobre transmissão quiral. Especialmente importante é a chamada organocatálise de reações orgânicas por prolina, por exemplo em reações de Mannich.
Alguns modelos propostos para a transmissão da assimetria quiral são a polimerização,[38][39][40][41][42][43] epimerização [44][45] ou copolimerização.[46][47]
Resolução óptica em aminoácidos racêmicos
Não existe nenhuma teoria que elucide as correlações entre L-aminoácidos. Se tomarmos, por exemplo, alanina, o qual tem um pequeno grupo metil, e fenilalanina, a qual tem um grupo benzil maior, uma pergunta simples é em que aspecto, L-alanina assemelha-se L-fenilalanina mais que D-fenilalanina e que tipo de mecanismo causa a seleção de todos L-aminoácidos, porque pode ser possível que a alanina tenha sido L e fenilalanina era D.
Foi relatado[48] em 2004 que excesso racêmico D,L-asparagina (Asn), o qual forma espontaneamente cristais de qualquer isômero durante a recristalização, induz resolução assimétrica de um aminoácido racêmico coexistente, tal como arginina (Arg), ácido aspártico (Asp), glutamina (Gln), histidina (His), leucina (Leu), metionina (Met), fenilalanina (Phe), serina (Ser), valina (Val), tirosina (Tyr) e triptofano (Trp). O excesso enantiomérico ee = 100 ×(L-D)/(L+D) desses aminoácidos foi correlacionado quase linearmente com o do indutor, i.e., Asn. Quando recristalizações de uma mistura de 12 D,L-aminoácidos (Ala, Asp, Arg, Glu, Gln, His, Leu, Met, Ser, Val, Phe, e Tyr) e excessos D,L-Asn foram feitos, todos os aminoácidos com a mesma configuração com Asn foram preferencialmente co-cristalizadas.[48] Foi incidental se o enriquecimento ocorreu em L- ou D-Asn, no entanto, uma vez feita a seleção, o aminoácido coexistente com a mesma configuração no carbono α foi preferencialmente envolvido devido à estabilidade termodinâmica na formação de cristais. O ee máximo foi relatado como sendo 100%. Com base nesses resultados, propõe-se que uma mistura de aminoácidos racêmicos causa resolução óptica espontânea e eficaz, mesmo que a síntese assimétrica de um único aminoácido não ocorra sem o auxílio de uma molécula opticamente ativa.
Este é o primeiro estudo que elucida razoavelmente a formação de quiralidade a partir de aminoácidos racêmicos com evidências experimentais.
História do termo
Este termo foi introduzido por Lorde Kelvin em 1904, ano em que publicou sua Palestra de Baltimore de 1884. Kelvin usou o termo homoquiralidade como uma relação entre duas moléculas, ou seja, duas moléculas são homoquirais se têm a mesma quiralidade.[35][49] Recentemente, no entanto, homoquiral tem sido usado no mesmo sentido que enantiomericamente puro. Isso é permitido em alguns periódicos (mas não encorajado),[50]:342[51] seu significado mudando para a preferência de um processo ou sistema por um único isômero óptico em um par de isômeros nesses periódicos.
Ver também
Referências
- ↑ Nelson, Lehninger; et al. (2008). Lehninger Principles of Biochemistry. [S.l.]: Macmillan. 474 páginas
- ↑ Cintas P. and Viedma C. (2012), On the Physical Basis of Asymmetry and Homochirality, Chirality. DOI: 10.1002/chir.22028
- ↑ Carroll, James D. (março 2009). «A new definition of life». Chirality. 21 (3): 354–358. PMID 18571800. doi:10.1002/chir.20590
- ↑ a b Julian, Ryan R.; Myung, Sunnie; Clemmer, David E. (janeiro 2005). «Do Homochiral Aggregates Have an Entropic Advantage?». The Journal of Physical Chemistry B. 109 (1): 440–444. PMID 16851034. doi:10.1021/jp046478x
- ↑ a b c Jafarpour, Farshid; Biancalani, Tommaso; Goldenfeld, Nigel (2017). «Noise-induced symmetry breaking far from equilibrium and the emergence of biological homochirality». Physical Review E. 95 (3). 032407 páginas. Bibcode:2017PhRvE..95c2407J. PMID 28415353. doi:10.1103/PhysRevE.95.032407
- ↑ Reusch, William. «Peptides & Proteins». Natural Products. Michigan State University. Consultado em 8 maio 2018
- ↑ Lam, Eric (1997). «Nucleic acids and proteins». In: Dey, P.M.; Harborne, J.B. Plant Biochemistry. Burlington: [s.n.] 315 páginas. ISBN 9780080525723
- ↑ Zubay, Geoffrey (2000). Origins of Life: On Earth and in the Cosmos. [S.l.]: Elsevier. 96 páginas. ISBN 9780080497617
- ↑ a b Seckbach, Joseph, ed. (2012). Genesis - in the beginning : precursors of life, chemical models and early biological evolution. Dordrecht: Springer. ISBN 9789400729407
- ↑ a b c Hazen, Robert M. (2007). Genesis : the scientific quest for life's origins. Washington, D.C.: Joseph Henry. ISBN 9780309103107
- ↑ Smith, Silas (julho 2009). «Chiral Toxicology: It's the Same Thing... Only Different». Toxicological Sciences. 110 (1): 4–30. PMID 19414517. doi:10.1093/toxsci/kfp097
- ↑ a b Meierhenrich, Uwe (2008). Amino acids and the asymmetry of life caught in the act of formation. Berlin: Springer. ISBN 9783540768869
- ↑ Shaw, Andrew M. (2007). Astrochemistry From Astronomy to Astrobiology. Chichester: John Wiley & Sons. 247 páginas. ISBN 9780470091388
- ↑ Guijarro, A. and Yus, M. The Origin of Chirality in the Molecules of Life (RSC Publishing, Cambridge, 2009), 1st ed.
- ↑ Chen Y, Ma W. The origin of biological homochirality along with the origin of life. PLoS Comput Biol. 2020 Jan 8;16(1):e1007592. doi: 10.1371/journal.pcbi.1007592. PMID: 31914131; PMCID: PMC6974302.
- ↑ Weller, M. G. (2024). The Mystery of Homochirality on Earth. Life, 14(3), 341. doi: 10.3390/life14030341
- ↑ Jaakkola, S; Sharma, V; Annila, A (2008). «Cause of chirality consensus». Curr. Chem. Biol. 2 (2): 53–58. arXiv:0906.0254. doi:10.2174/187231308784220536
- ↑ Globus, Noemie; Blandford, Roger D. (20 maio 2020). «The Chiral Puzzle of Life». The Astrophysical Journal Letters. 895 (1): L11. Bibcode:2020ApJ...895L..11G. arXiv:2002.12138. doi:10.3847/2041-8213/ab8dc6
- ↑ Barron, L. D. (1 de setembro de 1986). «True and false chirality and absolute asymmetric synthesis». Journal of the American Chemical Society. 108 (18): 5539–5542. Bibcode:1986JAChS.108.5539B. ISSN 0002-7863. doi:10.1021/ja00278a029
- ↑ Barron, L. D. (20 de agosto de 1981). «Optical activity and time reversal». Molecular Physics. 43 (6): 1395–1406. Bibcode:1981MolPh..43.1395B. ISSN 0026-8976. doi:10.1080/00268978100102151
- ↑ Clark, Stuart (Julho–agosto 1999). «Polarized Starlight and the Handedness of Life». American Scientist. 87 (4). 336 páginas. Bibcode:1999AmSci..87..336C. ISSN 0003-0996. doi:10.1511/1999.4.336
- ↑ Helman, Daniel S (6 julho 2018). «Galactic Distribution of Chirality Sources of Organic Molecules». Acta Astronautica. 151: 595–602. Bibcode:2018AcAau.151..595H. ISSN 0094-5765. arXiv:1612.06720. doi:10.1016/j.actaastro.2018.07.008
- ↑ Mislow, Kurt (2003). «Absolute Asymmetric Synthesis: A Commentary». Collection of Czechoslovak Chemical Communications (em inglês). 68 (5): 849–864. ISSN 1212-6950. doi:10.1135/cccc20030849
- ↑ Frank, F.C. (1953). «On spontaneous asymmetric synthesis». Biochimica et Biophysica Acta. 11 (4): 459–463. PMID 13105666. doi:10.1016/0006-3002(53)90082-1
- ↑ Note que em seu artigo original, Frank não propôs nenhum conjunto de reações químicas, mas um conjunto de equações dinâmicas, onde as concentrações de ambos os enantiômeros foram denotadas como [n1] e [n2] respectivamente.
- ↑ Jafarpour, Farshid; Biancalani, Tommaso; Goldenfeld, Nigel (2015). «Noise-induced mechanism for biological homochirality of early life self-replicators». Physical Review Letters. 115 (15). 158101 páginas. Bibcode:2015PhRvL.115o8101J. PMID 26550754. arXiv:1507.00044. doi:10.1103/PhysRevLett.115.158101
- ↑ Shibata, Takanori; Morioka, Hiroshi; Hayase, Tadakatsu; et al. (17 janeiro 1996). «Highly Enantioselective Catalytic Asymmetric Automultiplication of Chiral Pyrimidyl Alcohol». Journal of the American Chemical Society. 118 (2): 471–472. Bibcode:1996JAChS.118..471S. ISSN 0002-7863. doi:10.1021/ja953066g
- ↑ Soai, Kenso; Sato, Itaru; Shibata, Takanori (2001). «Asymmetric autocatalysis and the origin of chiral homogeneity in organic compounds». The Chemical Record. 1 (4): 321–332. ISSN 1528-0691. PMID 11893072. doi:10.1002/tcr.1017
- ↑ Shibata, Takanori; Morioka, Hiroshi; Hayase, Tadakatsu; Choji, Kaori; Soai, Kenso (1996). «Highly Enantioselective Catalytic Asymmetric Automultiplication of Chiral Pyrimidyl Alcohol». J. Am. Chem. Soc. 118 (2): 471–472. Bibcode:1996JAChS.118..471S. doi:10.1021/ja953066g
- ↑ Mathew, Suju P; Iwamura, Hiroshi; Blackmond, Donna G (21 junho 2004). «Amplification of Enantiomeric Excess in a Proline-Mediated Reaction». Angewandte Chemie International Edition. 43 (25): 3317–3321. PMID 15213963. doi:10.1002/anie.200453997
- ↑ Cooks, R. G.; Zhang, D.; Koch, K. J. (2001). «Chiroselective Self-Directed Octamerization of Serine: Implications for Homochirogenesis». Anal. Chem. 73 (15): 3646–3655. PMID 11510829. doi:10.1021/ac010284l
- ↑ Nanita, S.; Cooks, R. G. (2006). «Serine Octamers: Cluster Formation, Reactions, and Implications for Biomolecule Homochirality». Angew. Chem. Int. Ed. 45 (4): 554–569. PMID 16404754. doi:10.1002/anie.200501328
- ↑ Blackmond, Donna G.; Klussmann, Martin (2007). «Spoilt for choice: assessing phase behaviour models for the evolution of homochirality». Chem. Commun. (39): 3990–3996. PMID 17912393. doi:10.1039/b709314b
- ↑ Fletcher, Stephen P.; Jagt, Richard B. C.; Feringa, Ben L. (2007). «An astrophysically relevant mechanism for amino acid enantiomer enrichment». Chem. Commun. 2007 Jul 7 (25): 2578–2580. PMID 17579743. doi:10.1039/b702882b
- ↑ a b Córdova, Armando; Engqvist, Magnus; Ibrahem, Ismail; Casas, Jesús; Sundén, Henrik (2005). «Plausible origins of homochirality in the amino acid catalyzed neogenesis of carbohydrates». Chem. Commun. 15 (15): 2047–2049. PMID 15834501. doi:10.1039/b500589b
- ↑ Kondepudi, D. K.; Kaufman, R. J.; Singh, N. (1990). «Chiral Symmetry Breaking in Sodium Chlorate Crystallization». Science. 250 (4983): 975–976. Bibcode:1990Sci...250..975K. PMID 17746924. doi:10.1126/science.250.4983.975
- ↑ Noorduin, Wim L.; Izumi, Toshiko; Millemaggi, Alessia; Leeman, Michel; Meekes, Hugo; Van Enckevort, Willem J. P.; Kellogg, Richard M.; Kaptein, Bernard; Vlieg, Elias; Blackmond, Donna G. (janeiro 2008). «Emergence of a Single Solid Chiral State from a Nearly Racemic Amino Acid Derivative» (PDF). Journal of the American Chemical Society. 130 (4): 1158–1159. Bibcode:2008JAChS.130.1158N. PMID 18173274. doi:10.1021/ja7106349
- ↑ Sandars, P. G. H. (2003). «A toy model for the generation of homochirality during polymerization». Origins of Life and Evolution of the Biosphere. 33 (6): 575–587. Bibcode:2003OLEB...33..575S. ISSN 0169-6149. PMID 14601927. doi:10.1023/a:1025705401769
- ↑ Brandenburg, Axel; Multamäki, Tuomas (julho 2004). «How long can left and right handed life forms coexist?». International Journal of Astrobiology. 3 (3): 209–219. Bibcode:2004IJAsB...3..209B. ISSN 1473-5504. arXiv:q-bio/0407008. doi:10.1017/s1473550404001983
- ↑ Brandenburg, A.; Andersen, A. C.; Höfner, S.; Nilsson, M. (junho 2005). «Homochiral Growth Through Enantiomeric Cross-Inhibition». Origins of Life and Evolution of Biospheres. 35 (3): 225–241. Bibcode:2005OLEB...35..225B. ISSN 0169-6149. PMID 16228640. arXiv:q-bio/0401036. doi:10.1007/s11084-005-0656-9
- ↑ Wattis, Jonathan A. D.; Coveney, Peter V. (junho 2005). «Symmetry-breaking in Chiral Polymerisation». Origins of Life and Evolution of Biospheres. 35 (3): 243–273. Bibcode:2005OLEB...35..243W. ISSN 0169-6149. PMID 16228641. arXiv:physics/0402091. doi:10.1007/s11084-005-0658-7
- ↑ Saito, Yukio; Hyuga, Hiroyuki (15 de maio de 2005). «Chirality Selection in Open Flow Systems and in Polymerization». Journal of the Physical Society of Japan. 74 (5): 1629–1635. Bibcode:2005JPSJ...74.1629S. ISSN 0031-9015. arXiv:physics/0503057. doi:10.1143/jpsj.74.1629
- ↑ Blanco, Celia; Hochberg, David (2011). «Chiral polymerization: symmetry breaking and entropy production in closed systems». Phys. Chem. Chem. Phys. 13 (3): 839–849. Bibcode:2011PCCP...13..839B. ISSN 1463-9076. PMID 21057681. arXiv:1104.2225. doi:10.1039/c0cp00992j
- ↑ Plasson, R.; Bersini, H.; Commeyas, A. (17 de novembro de 2004). «Recycling Frank: Spontaneous emergence of homochirality in noncatalytic systems». Proceedings of the National Academy of Sciences. 101 (48): 16733–16738. Bibcode:2004PNAS..10116733P. ISSN 0027-8424. PMC 534711. PMID 15548617. doi:10.1073/pnas.0405293101
- ↑ Stich, Michael; Blanco, Celia; Hochberg, David (2013). «Chiral and chemical oscillations in a simple dimerization model». Phys. Chem. Chem. Phys. 15 (1): 255–261. Bibcode:2013PCCP...15..255S. ISSN 1463-9076. PMID 23064600. arXiv:1210.1872. doi:10.1039/c2cp42620j
- ↑ Wattis, Jonathan A. D.; Coveney, Peter V. (agosto 2007). «Sequence Selection during Copolymerization». The Journal of Physical Chemistry B. 111 (32): 9546–9562. ISSN 1520-6106. PMID 17658787. doi:10.1021/jp071767h
- ↑ Blanco, Celia; Hochberg, David (2012). «Homochiral oligopeptides by chiral amplification: interpretation of experimental data with a copolymerization model». Physical Chemistry Chemical Physics. 14 (7): 2301–11. Bibcode:2012PCCP...14.2301B. ISSN 1463-9076. PMID 22237639. arXiv:1202.2268. doi:10.1039/c2cp22813k
- ↑ a b Kojo, S.; Uchino, H.; Yoshimura, M.; Tanaka, K. (2004). «Racemic D,L-asparagine causes enantiomeric excess of other coexisting racemic D,L-amino acids during recrystallization: a hypothesis accounting for the origin of L-amino acids in the biosphere». Chem. Comm. (19): 2146–2147. PMID 15467844. doi:10.1039/b409941a
- ↑ Morris, David G. (2001). Stereochemistry. Cambridge: Royal Society of Chemistry. p. 30. ISBN 978-1-84755-194-8 Verifique o valor de
|url-access=limited(ajuda) - ↑ Anslyn, Eric V.; Dougherty, Dennis A. (2006). Modern physical organic chemistry. Sausalito, Calif.: University Science Books. ISBN 9781891389313
- ↑ No entanto, a mensagem pode ser confusa. Em Moss, G. P. (1 janeiro 1996). «Basic terminology of stereochemistry (IUPAC Recommendations 1996)» (PDF). Pure and Applied Chemistry. 68 (12): 2193–2222. doi:10.1351/pac199668122193. Consultado em 7 maio 2018, a entrada para Enantiomericamente puro/Enantiopuro diz "O uso de homoquiral como sinônimo é fortemente desencorajado"; mas a entrada para Homoquiral diz "Veja enantiomericamente puro/enantiopuro."