Glioma angiocêntrico
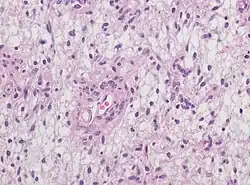
O glioma angiocêntrico (GA) refere-se a um tumor neuroepitelial raro em que as células malignas superficiais do cérebro envolvem os vasos cerebrais; é mais comummente encontrado em crianças e adultos jovens. Inicialmente identificado em 2005 pelo Dr. Ming-Tseh Wang e a sua equipa da Universidade do Texas em Austin, o GA foi classificado como um tumor de Grau I na Classificação de Tumores do Sistema Nervoso Central da OMS de 2007 devido ao seu comportamento clínico benigno, baixo índice de proliferação e propriedades curativas.[2] O GA afeta principalmente crianças e adultos jovens com uma idade média de diagnóstico inicial de 16 anos. Mais de 85% dos pacientes com GA sofrem de crises convulsivas intratáveis desde a infância, especialmente epilepsia parcial.[3]
Devido à sua curta história de 15 anos, à raridade da sua ocorrência e à falta de ensaios clínicos suficientes, o GA permanece difícil de compreender quanto aos sintomas, tratamentos e acompanhamento a longo prazo. Até agora, os cientistas e investigadores não encontraram a etiologia exata, testes patológicos definitivos para identificação, nem o efeito da radioterapia ou quimioterapia neste raro glioma indolente. No entanto, uma série de causas suspeitas está em discussão, incluindo a possível teoria da fusão proteica MYB-QKI na etiologia do GA. Atualmente, as ferramentas de diagnóstico padrão são a RM (Ressonância Magnética) e a TC (Tomografia Computorizada). Em termos de terapia, os pacientes são frequentemente submetidos a resseção subtotal ou total para remover a lesão problemática e têm uma probabilidade relativamente elevada de cura da doença. Contudo, ainda necessitam de períodos de acompanhamento mais extensos após a cirurgia para monitorização da recorrência tumoral e garantia de ausência de crises.
História
Antes da identificação do GA, os tipos mais frequentes e conhecidos de tumores glioneuronais raros no cérebro eram os tumores neuroepiteliais disembrioplásicos e os gangliogliomas.[4] Em 2002, Wang e o seu grupo da Universidade do Texas reconheceram um novo padrão neurológico em três casos de tumores hemisféricos cerebrais e publicaram pela primeira vez este subtipo raro de tumor GA na American Association of Neuropathologists.[5] Em 2005, Lellouch-Tubiana e a sua equipa analisaram histologicamente 204 espécimes de cirurgia de epilepsia de pacientes com idades entre os 2 e os 14 anos e descobriram três casos muito semelhantes aos mostrados por Wang et al. em termos de aparência patológica e resultados de RM.[4] O GA manifesta um padrão neuronal arquitetural e características morfológicas distintas, que foram posteriormente caracterizadas como uma nova entidade clinicopatológica denominada "tumores neuroepiteliais angiocêntricos" dentro do espectro de tumores glioneuronais pediátricos.[4] Simultaneamente, no mesmo mês em 2005, Wang et al. também publicaram as suas descobertas adicionais de cinco casos de GA e indicaram as propriedades histológicas, imuno-histoquímicas e ultraestruturais específicas deste tumor de crescimento lento e produtor de convulsões.[5] Em 2007, a Quarta Edição da Organização Mundial da Saúde reconheceu o GA como uma nova entidade clinicopatológica e designou-o como um tumor de Grau I associado à epilepsia, com células bipolares monomórficas e um arranjo de crescimento perivascular único.[2] Em 2016, a OMS efetuou algumas alterações e classificou o GA, o glioma cordoide do terceiro ventrículo e o astroblastoma como "Outros Gliomas" de Grau I.[6]
Sinais e sintomas
Os pacientes costumam apresentar uma história de crises convulsivas intratáveis entre os 3 e os 14 anos de idade.[7] A gravidade da crise depende frequentemente da localização do tumor e não do seu tamanho, uma vez que as lesões superficiais localizadas nos lobos frontal e temporal desencadeiam frequentemente uma maior possibilidade epileptogénica do que os tumores mais profundos.[8] Os sintomas comuns também incluem cefaleia, deficiência visual, tonturas, dor de ouvido, paragem da fala, ataxia e paresia, predominantemente em crianças e adultos jovens.[9] Os sintomas dos gliomas de baixo grau e crescimento lento são mais epileptogénicos, enquanto os gliomas de alto grau manifestam sintomas relacionados com o aumento da pressão intracraniana.[10]
Causas
Muitos estudos sobre o GA priorizaram características neurorradiológicas, aspetos clínicos, fisiopatologia e tratamento cirúrgico desta doença rara. A citogénese do GA permanece incerta desde 2005.
Como em 2005, a primeira investigação publicada postulava que o ependimoma e variantes do astroblastoma poderiam contribuir para a formação do tumor.[5] No mesmo ano, sugeriu-se que o GA deriva possivelmente da transformação neoplásica de células da glia radial durante a migração neuronal.[4]
Fusão Proteica MYB-QKI
Em 2016, propôs-se que fusões de genes que ativam a via MAPK podem induzir células malignas de GA.[11] Após análise de 172 subtipos de gliomas pediátricos, reconheceu-se a fusão proteica dos genes MYB e QKI na maioria dos perfis de GA. O MYB é um proto-oncogene, enquanto o QKI é um gene supressor de tumor.
Baixa recorrência após cirurgia
O GA comporta-se frequentemente como uma neoplasia indolente e é curável após resseção. Visto que o GA não adquire mutações da IDH1, possui um menor potencial de recorrência após a cirurgia em comparação com gliomas de graus II e III e glioblastomas secundários.[3]
Diagnóstico
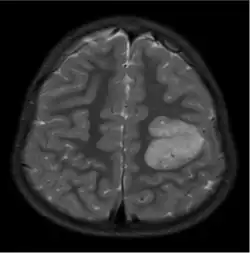
As características do GA são visíveis através de TC ou RM. Na RM, o GA aparece como lesões corticais bem delineadas, sólidas e hiperintensas em T2.[1] Outro traço diagnóstico é uma extensão para os ventrículos cerebrais adjacentes.[13]
O diagnóstico preciso de GA em relação a outros gliomas (como o astroblastoma ou o ganglioglioma) é um desafio meramente baseado em exames de imagem.[14]
Para confirmação, os clínicos requerem biopsia e coloração imuno-histoquímica. As células infiltrativas apresentam resultados positivos para a proteína gliofibrilar ácida (GFAP) e para o antigénio de membrana epitelial (EMA).[1] Outros marcadores incluem Ki-67, NeuN, proteína 53, sinaptofisina, Olig-2 e creatina quinase.[3]
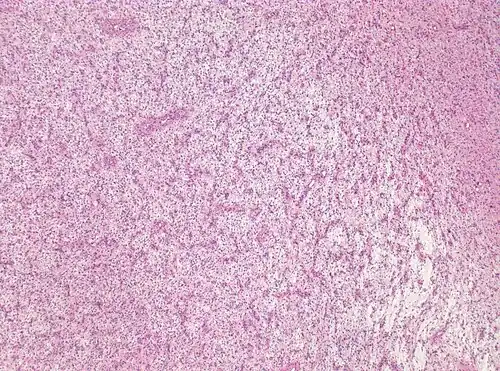
Tratamento
O tratamento definitivo é a resseção cirúrgica. Quase todos os pacientes escolhem a resseção subtotal ou total.[14] Uma resseção total consegue regredir o crescimento epileptogénico e curar esta neoplasia.[14] A resseção subtotal mostra uma taxa de recorrência mais elevada.[14] O prognóstico é consideravelmente favorável. A eficácia da quimioterapia ou radiação ainda não é conhecida para este tumor de baixo grau.
Epidemiologia
Até junho de 2020, os casos relatados de GA atingiram os 108 desde o relatório inicial de Wang et al. Dentro dos casos relatados, a patologia ocorre principalmente em crianças e adultos jovens.[3] A idade média do diagnóstico inicial é de 16 anos, e a prevalência em homens para mulheres é de uma proporção de 1,5 para 1.[3] O intervalo de diagnóstico inicial varia entre os 1,5 e os 83 anos, com uma mediana de 13.[3]
Dos 108 casos relatados, 102 apresentavam tumores GA numa localização supratentorial sob o córtex cerebral (94,4%), e 88 em 108 foram encontrados num único lobo (81,5%).[3] Foram registados 46 casos no lobo esquerdo do cérebro e 43 no lobo direito.[3] A localização mais comum onde o GA começa a crescer é o lobo temporal, com 39% dos casos relatados, seguido pelos lobos parietal (30%) e frontal (15%).[9] O local de crescimento menos comum é o tálamo, com apenas 1% dos casos relatados, e apenas foram descritos seis casos de lesões no tronco cerebral.[8]
94 dos 108 pacientes (87%) optaram por realizar algum grau de remoção cirúrgica: 61 pacientes foram submetidos a resseção total macroscópica (GTR, 64,9%) e 26 adotaram a resseção subtotal (STR, 27,7%).[3] O resultado da resseção foi altamente ideal, com 93,1% dos pacientes completamente livres de crises convulsivas durante o período de acompanhamento.[3] Os restantes seis pacientes que optaram por realizar STR experienciaram algum grau de recorrência dos sintomas.[3] Isto indica que o GA tem um prognóstico favorável, com uma taxa de mortalidade e de recorrência comparativamente baixas.
Referências
- ↑ a b c Shakur SF, McGirt MJ, Johnson MW, Burger PC, Ahn E, Carson BS, Jallo GI (Março 2009). «Angiocentric glioma: a case series». Journal of Neurosurgery. Pediatrics (em inglês). 3 (3): 197–202. PMC 2675755
 . PMID 19338465. doi:10.3171/2008.11.PEDS0858
. PMID 19338465. doi:10.3171/2008.11.PEDS0858
- ↑ a b Brat DJ, Scheithauer BW, Fuller GN, Tihan T (Julho 2007). «Newly codified glial neoplasms of the 2007 WHO Classification of Tumours of the Central Nervous System: angiocentric glioma, pilomyxoid astrocytoma and pituicytoma». Brain Pathology. 17 (3): 319–324. PMC 8095654. PMID 17598825. doi:10.1111/j.1750-3639.2007.00082.x
- ↑ a b c d e f g h i j k l Han G, Zhang J, Ma Y, Gui Q, Yin S (Agosto 2020). «Clinical characteristics, treatment and prognosis of angiocentric glioma». Oncology Letters. 20 (2): 1641–1648. PMC 7377082. PMID 32724405. doi:10.3892/ol.2020.11723
- ↑ a b c d Lellouch-Tubiana A, Boddaert N, Bourgeois M, Fohlen M, Jouvet A, Delalande O, et al. (Outubro 2005). «Angiocentric neuroepithelial tumor (ANET): a new epilepsy-related clinicopathological entity with distinctive MRI». Brain Pathology. 15 (4): 281–286. PMC 8095937. PMID 16389940. doi:10.1111/j.1750-3639.2005.tb00112.x
- ↑ a b c Wang M, Tihan T, Rojiani AM, Bodhireddy SR, Prayson RA, Iacuone JJ, et al. (Outubro 2005). «Monomorphous angiocentric glioma: a distinctive epileptogenic neoplasm with features of infiltrating astrocytoma and ependymoma». Journal of Neuropathology and Experimental Neurology. 64 (10): 875–881. PMID 16215459. doi:10.1097/01.jnen.0000182981.02355.10
- ↑ Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. (Junho 2016). «The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary». Acta Neuropathologica. 131 (6): 803–820. PMID 27157931. doi:10.1007/s00401-016-1545-1
- ↑ Preusser M, Hoischen A, Novak K, Czech T, Prayer D, Hainfellner JA, et al. (Novembro 2007). «Angiocentric glioma: report of clinico-pathologic and genetic findings in 8 cases». The American Journal of Surgical Pathology. 31 (11): 1709–1718. PMID 18059228. doi:10.1097/PAS.0b013e31804a7ebb
- ↑ a b da Silva JF, de Souza Machado GH, Pedro MK, Vosgerau R, Hunhevicz SC, Ramina R (Dez 2019). «Angiocentric glioma: Literature review and first case in Brazil». Interdisciplinary Neurosurgery. 18. 100508 páginas. ISSN 2214-7519. doi:10.1016/j.inat.2019.100508
- ↑ a b Alexandru D, Haghighi B, Muhonen MG (1 de março de 2013). «The treatment of angiocentric glioma: case report and literature review». The Permanente Journal. 17 (1): e100–e102. PMC 3627792. PMID 23596378. doi:10.7812/tpp/12-060
- ↑ Ampie L, Choy W, DiDomenico JD, Lamano JB, Williams CK, Kesavabhotla K, et al. (Junho 2016). «Clinical attributes and surgical outcomes of angiocentric gliomas». Journal of Clinical Neuroscience. 28: 117–122. PMID 26778052. doi:10.1016/j.jocn.2015.11.015
- ↑ Bandopadhayay P, Ramkissoon LA, Jain P, Bergthold G, Wala J, Zeid R, et al. (Março 2016). «MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism». Nature Genetics. 48 (3): 273–282. PMC 4767685. PMID 26829751. doi:10.1038/ng.3500
- ↑ Kumar M, Ramakrishnaiah R, Samant R (2013). «Angiocentric glioma, a recently added WHO grade-I tumor». Radiology Case Reports. 8 (4). 782 páginas. PMC 4899553. PMID 27330646. doi:10.2484/rcr.v8i4.782
- ↑ Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2016). WHO classification of tumours of the central nervous system Revised 4th ed. Lyon: World Health Organization, International Agency for Research on Cancer. ISBN 978-92-832-4492-9. OCLC 951745876
- ↑ a b c d Gatto L, Franceschi E, Nunno VD, Tomasello C, Bartolini S, Brandes AA (3 de julho de 2020). «Glioneuronal tumors: clinicopathological findings and treatment options». Future Neurology. 15 (3): FNL47. ISSN 1479-6708. doi:10.2217/fnl-2020-0003