Síndrome de Hanhart
| Síndrome de Hanhart | |
|---|---|
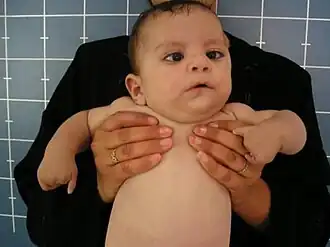 | |
| Um bebê com síndrome de Moebius e síndrome de hipogênese oromandibular e de membros, exibindo sintomas semelhantes aos da síndrome de Hanhart. | |
| Sintomas | Subdesenvolvimento da boca, mandíbula, língua e extremidades |
| Início habitual | Neonatal, pré-natal |
| Duração | Ao longo de toda a vida |
| Causas | Distúrbios vasculares durante o desenvolvimento fetal |
| Condições semelhantes | Disostose acrofacial de Nager [en]
Síndrome de Johnson Hall Krous Síndrome de Goldenhar |
| Frequência | <1/1.000.000, 30 casos conhecidos |
| Classificação e recursos externos | |
| CID-10 | Q87.2 |
| CID-9 | 759.89 |
| OMIM | 103300 |
| MeSH | C535629 |
A síndrome de Hanhart[a] é uma condição médica amplamente classificada que envolve distúrbios congênitos caracterizados por uma língua subdesenvolvida e malformações em extremidades e dedos. Existem cinco tipos da síndrome, com a gravidade e a natureza da condição variando amplamente de caso para caso. A síndrome de Hanhart é classificada como uma doença rara, com aproximadamente 30 casos conhecidos relatados entre 1932 e 1991. Hipóteses iniciais sugeriam que a condição era causada por fatores genéticos, enquanto uma hipótese mais recente indica que pode ser resultado de lesões hemorrágicas durante o desenvolvimento pré-natal. O mecanismo causal por trás dessa disrupção vascular ainda é desconhecido.
Descoberta e etimologia
A síndrome de Hanhart foi descrita pela primeira vez em 1932 por Ernst Hanhart. O termo em inglês, "Hanhart syndrome", só foi utilizado em 1950, quando Hanhart descreveu três pacientes nascidos com defeitos nos membros e ausência de língua.[1] Em 1971, a síndrome foi classificada de forma mais ampla como síndromes de hipogênese oromandibular e de membros, abrangendo uma gama de distúrbios que compartilham microglossia. O nome em inglês, hypoglossia-hypodactylia syndrome, foi proposto em 1971 como uma designação mais precisa, sendo usado como sinônimo.[2][3] Devido à ampla variedade de sintomas, a síndrome recebeu diversos nomes ao longo de sua história diagnóstica.[4]
Fisiopatologia
A síndrome de Hanhart faz parte da família de condições conhecidas como síndrome de hipogênese oromandibular e de membros (SHOM), caracterizadas pelo subdesenvolvimento da boca, mandíbula, língua e extremidades.[2][1] A síndrome é marcada por uma língua subdesenvolvida (microglossia), boca pequena (microstomia [en]), mandíbula menor que a média (micrognatismo), fissura ou fixação anormal da língua, ausência de dentes (hipodontia mandibular), fenda labial e palatina, paralisias de nervos cranianos, incluindo a síndrome de Moebius, nariz largo, maior distância entre os olhos (telecanto [en]), defeitos nas pálpebras inferiores e assimetria facial.[2][1][5]
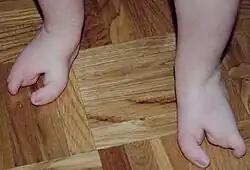
Os membros geralmente apresentam subdesenvolvimento (hipoplasia), que pode variar desde a ausência de falanges (hipodactilia) até adactilia parcial, amputação parcial ou malformação dos membros (peromelia). Quando há defeitos nos membros, geralmente afetam os quatro.[5] A perda completa da língua (aglossia) e dos dedos (adactilia) não foi relatada.[3] Sintomas menos comuns incluem gastrosquise e fusão anormal do baço com as gônadas (fusão esplenogonadal [en]).[5] Mamilos supranumerários, microcefalia e micropênis também foram identificados como possíveis sintomas.[6] Exames de ultrassonografia das regiões cranial e abdominal em pacientes mostram que as estruturas internas não são afetadas pela síndrome.[7] Deficiências mentais são raras, mas dificuldades de alimentação e fala foram relatadas em alguns casos.[5] A gravidade dessas deficiências varia muito entre os pacientes, que frequentemente apresentam alguns, mas não todos, os sintomas.[2][1]
A síndrome de Hanhart é classificada em cinco tipos, que indicam os sintomas e a gravidade da condição. O Tipo I envolve hipoglossia e aglossia parcial sem outros sintomas. O Tipo II inclui hipoglossia com hipomelia ou hipodactilia. O Tipo III abrange hipoglossia com anquilose e hipomelia ou hipodactilia. O Tipo IV apresenta hipoglossia ou hipomelia/hipodactilia. O Tipo V combina várias síndromes comórbidas com a síndrome de Hanhart, incluindo síndrome de Pierre Robin, síndrome de Moebius e síndrome da banda de constrição [en].[3][8]
Causas e prevalência
As causas da síndrome de Hanhart e outras condições de SHOM são desconhecidas, mas fatores genéticos e ambientais foram propostos.[9] A síndrome segue um padrão de herança autossômica dominante, com prevalência populacional inferior a 1/1.000.000, sendo classificada como uma doença rara.[5][10] Entre 1932 e 1991, cerca de 30 casos foram relatados na literatura.[5]
A prevalência da síndrome em pacientes com consanguinidade levou a uma hipótese inicial de que uma mutação em um gene autossômico recessivo causava a condição.[5] Até o momento, nenhum gene específico foi identificado.[7] Em 1973, foi proposta a hipótese de que lesões hemorrágicas ou coágulos sanguíneos durante o desenvolvimento pré-natal seriam a causa. Essa hipótese sugere que lesões vasculares reduzem o fluxo sanguíneo para os membros, língua e, raramente, partes do cérebro, causando malformações nas áreas afetadas.[1][5] Estudos em animais indicam que disrupções vasculares na quarta semana embrionária podem resultar na síndrome, mas o mecanismo permanece desconhecido.[11] Interações entre as camadas germinativas ectoderme e mesoderme,[4] e o uso de cloridrato de meclizina durante a gravidez foram sugeridos como causas potenciais.[12] Também foi proposto que a síndrome de Poland e a síndrome de Hanhart podem ter causas semelhantes, representando um espectro de um distúrbio maior.[4]
O uso de biópsia das vilosidades coriónicas também foi identificado como um fator contribuinte para a patogênese. Quando realizada nas primeiras 10 semanas de amenorreia, a biópsia das vilosidades coriónicas aumenta a correlação com o risco de o feto desenvolver a síndrome de Hanhart. Além disso, a CVS pode causar danos vasculares ao feto, resultando em disgênese oromandibular de membros e outros distúrbios pré-natais, reforçando a hipótese de disrupção vascular.[13][5][1]
Diagnóstico e tratamentos
O diagnóstico da síndrome de Hanhart baseia-se na apresentação clínica.[11] O diagnóstico diferencial deve descartar a disostose acrofacial de Nager [en], a síndrome de Johnson-Hall-Krous e a síndrome de Goldenhar.[11][5] Essas síndromes são diferenciadas pelo tipo e gravidade das deformidades faciais e dos membros.[5]
Os tratamentos variam conforme os sintomas específicos de cada paciente.[2] Podem incluir cirurgias ortopédicas ou plásticas para corrigir deformidades nos membros.[5] Essas intervenções abrangem correções cirúrgicas da língua, boca e mandíbula, além do uso de próteses e fisioterapia. A terapia fonoaudiológica é comum para tratar deformidades faciais.[1] Durante cirurgias sob anestesia, cuidados especiais com as vias aéreas são frequentemente necessários devido às deformidades craniofaciais.[11]
Casos notáveis
Referências
- ↑ a b c d e f g «Hanhart Syndrome - National Organization for Rare Disorders». rarediseases.org (em inglês). Consultado em 25 de fevereiro de 2023. Cópia arquivada em 30 de setembro de 2021
- ↑ a b c d e «Hanhart syndrome - National Organization for Rare Disorders». rarediseases.org (em inglês). 16 de junho de 2022. Consultado em 25 de fevereiro de 2023. Cópia arquivada em 25 de fevereiro de 2023
- ↑ a b c McKusick VA, Kniffin CL (8 de agosto de 2016). «Hypoglossia-Hypodactylia». omim.org (em inglês). Consultado em 25 de fevereiro de 2023. Cópia arquivada em 17 de junho de 2022
- ↑ a b c Herrmann J, Pallister PD, Gilbert EF, Vieseskul C, Bersu E, Pettersen JC, Opitz JM (abril de 1976). «Studies of malformation syndromes of man XXXXI B: nosologic studies in the Hanhart and the Möbius syndrome». European Journal of Pediatrics. 122 (1): 19–55. PMID 1261566. doi:10.1007/BF00445030
- ↑ a b c d e f g h i j k l «Orphanet: Hypoglossia hypodactyly syndrome». www.orpha.net. Consultado em 25 de fevereiro de 2023. Cópia arquivada em 25 de fevereiro de 2023
- ↑ Gathwala G, Singh J, Dalal P, Garg A (março de 2011). «Hypoglossia-hypodactyly syndrome in a newborn». Journal of Cranio-Maxillo-Facial Surgery. 39 (2): 99–101. PMID 20673638. doi:10.1016/j.jcms.2010.06.007
- ↑ a b Varal IG, Dogan P (29 de abril de 2019). «Hanhart syndrome: hypoglossia-hypodactylia syndrome». Pan African Medical Journal (em inglês). 32 (1): 213. ISSN 1937-8688. PMC 6620079. PMID 31312325. Consultado em 10 de março de 2023. Cópia arquivada em 26 de fevereiro de 2023
- ↑ Goyal M, Singh A, Singh P, Kapoor S (abril de 2014). «Hypoglossia-hypodactyly syndrome with short stature - a case report». Journal of Clinical and Diagnostic Research. 8 (4): SD01–SD02. PMC 4064859. PMID 24959494. doi:10.7860/JCDR/2014/7809.4281 (inativo 1 de novembro de 2024)
- ↑ Bersu ET, Pettersen JC, Charboneau WJ, Opitz JM (abril de 1976). «Studies of malformation syndromes of man XXXXIA: anatomical studies in the Hanhart syndrome--a pathogenetic hypothesis». European Journal of Pediatrics. 122 (1): 1–17. PMID 1261565. doi:10.1007/BF00445029
- ↑ «Hanhart syndrome - About the Disease - Genetic and Rare Diseases Information Center». rarediseases.info.nih.gov (em inglês). Consultado em 25 de fevereiro de 2023. Cópia arquivada em 5 de janeiro de 2023
- ↑ a b c d Bissonnette, Bruno; Luginbuehl, Igor; Engelhardt, Thomas (2019), «Hypoglossia-Hypodactylia Syndrome» 2 ed. , New York, NY: McGraw-Hill Education, Syndromes: Rapid Recognition and Perioperative Implications, consultado em 27 de fevereiro de 2023, cópia arquivada em 10 de março de 2023
- ↑ Bökesoy I, Aksüyek C, Deniz E (Julho de 1983). «Oromandibular limb hypogenesis/Hanhart's syndrome: possible drug influence on the malformation». Clinical Genetics. 24 (1): 47–49. PMID 6616945. doi:10.1111/j.1399-0004.1983.tb00068.x
- ↑ Chen, C. P.; Liu, F. F.; Jan, S. W.; Lin, S. P.; Lan, C. C. (14 de junho de 1996). «CVS-exposed limb deficiency defects with or without other birth defects: presentation of six cases born during a period of nine years». American Journal of Medical Genetics. 63 (3): 447–453. ISSN 0148-7299. PMID 8737650. doi:10.1002/(SICI)1096-8628(19960614)63:3<447::AID-AJMG6>3.0.CO;2-O
- ↑ Petter O (20 de julho de 2017). «Bodybuilder with one arm and no legs becomes internet sensation». The Independent. Consultado em 19 de abril de 2018. Cópia arquivada em 19 de abril de 2018
- ↑ Stump S (outubro de 2016). «'Dead' man living: Disabled teen-turned-Internet star 'lives by inspiring others'». Today. Consultado em 19 de abril de 2018. Cópia arquivada em 19 de abril de 2018
Notas
- ↑ Também conhecida como aglossia-adactilia; síndrome de hipoglossia-hipodactilia; peromelia com micrognatia; e síndrome de Jussieu.