Síndrome de Loeys-Dietz
| Síndrome de Loeys-Dietz | |
|---|---|
| Sinónimos | Síndrome de aneurisma aórtico devido a anomalias dos receptores TGF-beta |
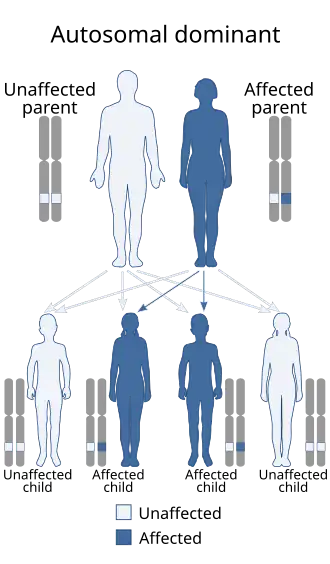 | |
| Esta condição é herdada de maneira autossômica dominante[1] | |
| Pronúncia | [ˌloʊiːzˈdiːts] LOH-eez-DEETS[2] |
| Especialidade | cardiologia, reumatologia, genética médica |
| Classificação e recursos externos | |
| CID-10 | Q87.4 |
| CID-9 | 759.89 |
| OMIM | 609192 |
| DiseasesDB | 34032 |
| MeSH | D055947 |
A síndrome de Loeys-Dietz (SLD) é uma doença genética autossômica dominante que afeta o tecido conjuntivo. Apresenta características semelhantes à síndrome de Marfan e à síndrome de Ehlers-Danlos.[3][4][5][6] A doença é caracterizada por aneurismas na aorta, frequentemente em crianças, e a aorta também pode sofrer dissecção súbita nas camadas enfraquecidas da parede da aorta.[7] Aneurismas e dissecções também podem ocorrer em artérias além da aorta.
Como os aneurismas em crianças tendem a se romper precocemente, as crianças estão em maior risco de morte se a síndrome não for identificada.[8] A cirurgia para reparar aneurismas aórticos é essencial para o tratamento. Anteriormente, acreditava-se que a expectativa de vida de um indivíduo com essa condição era de cerca de 30 a 40 anos, mas com avanços nos tratamentos, como possibilidades de cirurgia e medicamentos como o losartan, agora está comprovado que a expectativa de vida pode ser completa com a atenção médica adequada e exames regulares.
Existem cinco tipos da síndrome, designados de I a V, causados por mutações nos genes TGFBR1, TGFBR2, SMAD3, TGFB2 e TGFB3, respectivamente. Esses cinco genes que codificam fator de crescimento transformante desempenham um papel na sinalização celular que promove o crescimento e desenvolvimento dos tecidos do corpo.[9]
Mutações desses genes causam a produção de proteínas sem função. As células da pele de indivíduos com a síndrome de Loeys-Dietz não conseguem produzir colágeno, a proteína que confere força e elasticidade às células da pele.[8] Isso torna esses indivíduos suscetíveis a rupturas na pele, como hérnias. Embora a doença tenha um padrão de herança autossômico, ela resulta de uma nova mutação genética em 75% dos casos e ocorre em pessoas sem histórico da doença na família. Em outros casos, é herdada de um dos pais afetados.
A síndrome de Loeys-Dietz foi identificada e caracterizada pelos geneticistas pediátricos Bart Loeys e Harry "Hal" Dietz na Universidade Johns Hopkins em 2005.[7]
Sinais e sintomas
Há uma considerável variabilidade no fenótipo da síndrome de Loeys-Dietz, desde características leves até anormalidades sistêmicas graves. As principais manifestações da síndrome de Loeys-Dietz são tortuosidade arterial (curso sinuoso dos vasos sanguíneos), olhos amplamente espaçados (hipertelorismo), úvula larga ou bifurcada e aneurismas na raiz aórtica. Outras características podem incluir fenda palatina e uma aparência azul/acinzentada da esclera dos olhos. Defeitos cardíacos e pé torto podem ser observados ao nascimento.[10]
Há sobreposição nas manifestações das síndromes de Loeys-Dietz e Marfan, incluindo aumento do risco de aneurisma da aorta ascendente e dissecção aórtica, membros e dedos anormalmente longos, e ectasia dural (um estiramento e enfraquecimento gradual da dura-máter que pode causar dor abdominal e nas pernas). Achados como hipertelorismo (olhos amplamente espaçados), úvula bifurcada ou larga e achados cutâneos, como hematomas fáceis ou cicatrizes anormais, podem distinguir a síndrome de Loeys-Dietz da síndrome de Marfan.[6]
Indivíduos afetados frequentemente desenvolvem problemas relacionados ao sistema imunológico, como alergias alimentares, asma, rinite alérgica e transtornos inflamatórios, como eczema ou doença inflamatória intestinal.[6]
Os achados da síndrome de Loeys-Dietz podem incluir:
- Malformações esqueléticas
- Craniossinostose
- Escoliose
- Cifose
- Instabilidade espinhal e espondilolistese
- Anormalidades do esterno (pectus excavatum/pectus carinatum)
- Hipermobilidade articular
- Camptodactilia [en]
- Dedos longos
- Pé torto
- Anormalidades faciais
- Músculos oculares enfraquecidos ou ausentes (estrabismo)
- Olhos amplamente espaçados (hipertelorismo)
- Defeitos cardíacos congênitos
- Persistência do canal arterial
- Defeito septal atrial
- Válva aórtica bicúspide
- Artérias pulmonares cruzadas
- Translucidez da pele com textura aveludada
- Ma formação de Arnold-Chiari
Causa
Tipos (nomenclatura antiga)
Várias causas genéticas da síndrome de Loeys-Dietz foram identificadas. Uma mutação de novo no gene TGFB3, um ligante da via TGF-β, foi identificada em um indivíduo com uma síndrome que apresenta sintomas parcialmente sobrepostos aos da síndrome de Marfan e da síndrome de Loeys-Dietz.[11]
| Tipo | Gene | Locus | OMIM | Descrição |
|---|---|---|---|---|
| 1A | TGFBR1 | 9q22 | OMIM 609192 | Também conhecida como síndrome de Furlong |
| 1B | TGFBR2 | 3p22 | OMIM 610168 | - |
| 2A | TGFBR1 | 9q22 | OMIM 608967 | - |
| 2B | TGFBR2 | 3p22 | OMIM 610380 | Anteriormente conhecida como síndrome de Marfan tipo 2 |
| 3 | SMAD3 | 15q22.33 | OMIM 613795 | Também conhecida como síndrome de aneurismas-osteoartrite |
| 4 | TGFB2 | 1q41 | OMIM 614816 | - |
| 5 | TGFB3 | 14q24.3 | OMIM 615582 | - |
| 6 | SMAD2 | 18q21.1 | 619656 | - |
Diagnóstico
O diagnóstico envolve a consideração de características físicas e testes genéticos.[12] A presença de úvula bifurcada é uma característica diferenciadora da síndrome de Marfan, assim como a gravidade dos defeitos cardíacos. Pacientes com síndrome de Loeys-Dietz apresentam maior envolvimento cardíaco, e recomenda-se que sejam tratados para aorta aumentada mais cedo devido ao maior risco de ruptura precoce.
Como diferentes pessoas expressam diferentes combinações de sintomas e a síndrome foi identificada pela primeira vez em 2005, muitos médicos podem não estar cientes de sua existência.[13]
Tratamento
Como não há cura conhecida, a síndrome de Loeys-Dietz é uma condição vitalícia. Devido ao alto risco de morte por ruptura de aneurisma aórtico, os pacientes devem ser acompanhados de perto para monitorar a formação de aneurismas, que podem ser corrigidos com cirurgia vascular. Pesquisas anteriores em camundongos de laboratório sugeriram que o antagonista do receptor da angiotensina II losartan, que parece bloquear a atividade do TGF-beta, pode retardar ou interromper a formação de aneurismas aórticos na síndrome de Marfan. Um grande ensaio clínico patrocinado pelos National Institutes of Health está em andamento para explorar o uso de losartan na prevenção de aneurismas em pacientes com síndrome de Marfan. Tanto a síndrome de Marfan quanto a de Loeys-Dietz estão associadas ao aumento da sinalização de TGF-beta na parede dos vasos. Portanto, o losartan também é promissor para o tratamento da síndrome de Loeys-Dietz.[14] Em pacientes nos quais o losartan não interrompe o crescimento da aorta, o irbesartan demonstrou eficácia e está sendo estudado e prescrito para alguns pacientes com essa condição.
Se houver aumento da frequência cardíaca, um betabloqueador cardioseletivo beta-1, com ou sem losartan, é às vezes prescrito para reduzir a frequência cardíaca e evitar pressão adicional no tecido da aorta. Da mesma forma, atividades físicas extenuantes, como levantamento de peso e esportes de contato, são desencorajadas em pacientes.[15]
Referências
- ↑ «Sindrome de Loeys-Dietz». Orphanet (em inglês). Consultado em 1 de setembro de 2025. Arquivado do original em 30 de julho de 2017
- ↑ «Research and Treatment | Loeys-Dietz Syndrome». Johns Hopkins Medicine. 10 de agosto de 2018. Consultado em 1 de setembro de 2025. Arquivado do original em 19 de dezembro de 2021
- ↑ Loeys BL, Schwarze U, Holm T et al. (2006). «Aneurysm syndromes caused by mutations in the TGF-beta receptor» 8 ed. N. Engl. J. Med. 355: 788–98. PMID 16928994. doi:10.1056/NEJMoa055695
- ↑ LeMaire SA, Pannu H, Tran-Fadulu V, Carter SA, Coselli JS, Milewicz DM (2007). «Severe aortic and arterial aneurysms associated with a TGFBR2 mutation» 3 ed. Nature Clinical Practice Cardiovascular Medicine. 4: 167–71. PMC 2561071. PMID 17330129. doi:10.1038/ncpcardio0797
- ↑ Loeys BL, Chen J, Neptune ER et al. (março de 2005). «A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2» 3 ed. Nat. Genet. 37: 275–81. PMID 15731757. doi:10.1038/ng1511. hdl:1854/LU-330238
- ↑ a b c Meester, Josephina A. N.; Verstraeten, Aline; Schepers, Dorien; Alaerts, Maaike; Van Laer, Lut; Loeys, Bart L. (novembro de 2017). «Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome». Annals of Cardiothoracic Surgery (6): 582–594. PMC 5721110. PMID 29270370. doi:10.21037/acs.2017.11.03. Consultado em 29 de setembro de 2025
- ↑ a b Van Laer, Lut; Dietz, Harry; Loeys, Bart (2014). Halper, Jaroslava, ed. «Loeys-Dietz Syndrome». Dordrecht: Springer Netherlands (em inglês): 95–105. ISBN 978-94-007-7893-1. doi:10.1007/978-94-007-7893-1_7. Consultado em 29 de setembro de 2025
- ↑ a b Akazawa, Yohei; Motoki, Noriko; Tada, Akira; Yamazaki, Shoko; Hachiya, Akira; Matsuzaki, Satoshi; Kamiya, Motoko; Nakamura, Tomohiko; Kosho, Tomoki (2016). «Decreased Aortic Elasticity in Children With Marfan Syndrome or Loeys-Dietz Syndrome». Circulation Journal (11): 2369–2375. doi:10.1253/circj.CJ-16-0739. Consultado em 29 de setembro de 2025
- ↑ Fontana, Paolo; Genesio, Rita; Casertano, Alberto; Cappuccio, Gerarda; Mormile, Angela; Nitsch, Lucio; Iolascon, Achille; Andria, Generoso; Melis, Daniela (15 de março de 2014). «Loeys–Dietz syndrome type 4, caused by chromothripsis, involving the TGFB2 gene». Gene (1): 69–73. ISSN 0378-1119. doi:10.1016/j.gene.2014.01.017. Consultado em 29 de setembro de 2025
- ↑ «Sindrome de Loeys-Dietz». The Marfan Foundation (em inglês). 27 de junho de 2013. Consultado em 1 de setembro de 2025
- ↑ Rienhoff HY, Yeo C-Y, Morissette R, Khrebtukova I, Melnick J, Luo S, Leng N, Kim Y-J, Schroth G, Westwick J, Vogel H, McDonnell N, Hall JG, Whitman M. 2013. A mutation in TGFB3 associated with a syndrome of low muscle mass, growth retardation, distal arthrogryposis, and clinical features overlapping between Marfan and Loeys–Dietz syndrome. Am J Med Genet Part A. 161A:2040–2046.
- ↑ MacCarrick, Gretchen; Black, James H.; Bowdin, Sarah; El-Hamamsy, Ismail; Frischmeyer-Guerrerio, Pamela A.; Guerrerio, Anthony L.; Sponseller, Paul D.; Loeys, Bart; Dietz, Harry C. (1 de agosto de 2014). «Loeys–Dietz syndrome: a primer for diagnosis and management». Genetics in Medicine (em inglês) (8): 576–587. ISSN 1098-3600. PMC 4131122. PMID 24577266. doi:10.1038/gim.2014.11. Consultado em 29 de setembro de 2025
- ↑ Linde, Denise van der; Roos-Hesselink, Jolien; Loeys, Bart L. (3 de outubro de 2016). Aneurysms-Osteoarthritis Syndrome: SMAD3 Gene Mutations (em inglês). [S.l.]: Elsevier. Consultado em 29 de setembro de 2025
- ↑ Sandor, George G. S.; Alghamdi, Mohammed H.; Raffin, Leslie A.; Potts, Mary T.; Williams, Lindsey D.; Potts, James E.; Kiess, Marla; Breemen, Casey van (20 de janeiro de 2015). «A randomized, double blind pilot study to assess the effects of losartan vs. atenolol on the biophysical properties of the aorta in patients with Marfan and Loeys–Dietz syndromes». International Journal of Cardiology (em inglês): 470–475. ISSN 0167-5273. PMID 25465809. doi:10.1016/j.ijcard.2014.11.082. Consultado em 9 de outubro de 2025
- ↑ Loeys, BL; Dietz, HC (1993). «Sindrome de Loeys-Dietz». In: Adam, MP; Ardinger, HH; Pagon, RA. GeneReviews®. Seattle (WA): University of Washington, Seattle. PMID 20301312. Consultado em 1 de setembro de 2025
Leitura adicional
- Bertoli-Avella, A. M; Gillis, E; Morisaki, H; Verhagen, J. M. A; De Graaf, B. M; Van De Beek, G; Gallo, E; Kruithof, B. P. T; Venselaar, H; Myers, L. A; Laga, S; Doyle, A. J; Oswald, G; Van Cappellen, G. W. A; Yamanaka, I; Van Der Helm, R. M; Beverloo, B; De Klein, A; Pardo, L; Lammens, M; Evers, C; Devriendt, K; Dumoulein, M; Timmermans, J; Bruggenwirth, H. T; Verheijen, F; Rodrigus, I; Baynam, G; Kempers, M; et al. (2015). «Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections» 13 ed. Journal of the American College of Cardiology. 65: 1324–1336. PMC 4380321. PMID 25835445. doi:10.1016/j.jacc.2015.01.040