Galactose-1-fosfato uridililtransferase
- Para o tecido linfoide associado ao intestino ver GALT.
| Galactose-1-fosfato uridililtransferase | |
|---|---|
Dímero da galactose-1-fosfato uridiltransferase de Escherichia coli. |
A galactose-1-fosfato uridiltransferase (GALT) (EC 2.7.7.12) é uma enzima responsável por converter a galactose ingerida em glicose.[1]
A GALT catalisa a segunda etapa da rota de Leloir do metabolismo da galactose.

O enzima está codificado no gene GALT do cromossomo 9,[2] e a sua expressão é controlada pelo gene FOX3. A ausência deste enzima origina nos humanos a doença galactosemia que pode ser mortal nos neonatos se não se elimina da dieta a lactose (dissacarídeo que contém galactose). A fisiopatologia da galactosemia não foi ainda definida claramente.[1]
Mecanismo
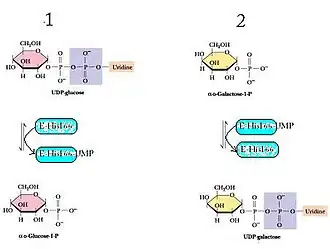
A galactose-1-fosfato uridiltransferase (GALT) catalisa a segunda reação da rota de Leloir do metabolismo da galactose por meio de uma cinética enzimática de duplo deslocamento.[4] Isto significa que a reação líquida consiste em dois reagentes e dois produtos, e procede por meio do seguinte mecanismo: O enzima reage com um substrato para gerar um produto e o enzima modificado, o qual posteriormente reage com o segundo substrato para formar o segundo produto, regenerando-se o enzima original.[5] No caso da GALT, o resíduo His-166 atua como um nucleófilo potente para facilitar a transferência de um nucleótido entre UDP-hexoses e hexoses-1-fosfato.[6]
- UDP-glicose + Enzima-His
 Glicose 1-fosfato + Enzima-His-UMP
Glicose 1-fosfato + Enzima-His-UMP - Galactose 1-fosfato + Enzima-His-UMP UDP-galactose + Enzima-His[6]
A GALT necessita de um ião ferro e um ião zinco por subunidade.[7]
Estudos estruturais
| Galactose-1-fosfato uridiltransferase, domínio N-terminal | |
|---|---|
| Indicadores | |
| Pfam | PF01087 |
| PROSITE | PDOC00108 |
| SCOP | 1hxp |
| Galactose-1-fosfato uridiltransferase, domínio C-terminal | |
|---|---|
| Indicadores | |
| Pfam | PF02744 |
| InterPro | IPR005850 |
| PROSITE | PDOC00108 |
| SCOP | 1hxp |
A GALT é um homodímero.[7] A estrutura tridimensional por cristalografia de raios X a uma resolução de 1,8 ángstroms da galactose-1-fosfato uridiltransferase foi obtida por Wedekind, Frey e Rayment, e a sua análise estrutural encontrou os aminoácidos chave essenciais para a função da GALT.[6] Os aminoácidos importantes encontrados por Wedekind et al. (Leu-4, Phe-75, Asn-77, Asp-78, Phe-79 e Val-108) são concordantes com os resíduos que foram implicados em experimentos de mutações pontuais e em rastreio clínico na galactosemia humana.[6][8]
Importância clínica
A deficiência em galactose-1-fosfato uridiltransferase causa a galactosemia clássica. A galactosemia é uma doença da infância de natureza hereditária.[9] Os traços autossómicos recessivos afetam aproximadamente 1 em cada 40.000-60.000 nascidos. A galactosemia clássica (G/G) é causada por uma deficiência na atividade da GALT, enquanto que a forma mais comum, Duarte/Clássica (D/G), se produz pela atenuação da atividade da GALT.[10] Os sintomas incluem problemas ováricos, dispraxia (dificuldade para falar correta e consistentemente)[11] e déficits neurológicos.[10] Uma única mutação nalguns aminoácidos pode produzir atenuação ou deficiência da atividade da GALT.[12] Por exemplo, uma mutação simples de A a G no éxon 6 do gene GALT muda o Glu-188 por Arg, e uma mutação de A a G no éxon 10 muda a Asn-314 por Asp.[10] Estas duas mutações também adicionam novos sítios de corte para as enzimas de restrição.[10] O rastreio clínico praticamente eliminou a morte neonatal pela galactosemia clássica, mas a doença, devido ao papel da GALT no metabolismo da conversão da galactose ingerida em glicose, pode ser mortal.[9][13] Contudo, as pessoas afetadas pela galactosemia podem viver de forma relativamente normal evitando os produtos que contêm leite e qualquer alimento que contenha galactose, ainda que haja um risco potencial para problemas no desenvolvimento neurológico, ou outras complicações, mesmo naqueles indivíduos que evitem a galactose.[14]
Referências
- ↑ a b «Entrez Gene: GALT galactose-1-phosphate uridylyltransferase»
- ↑ OMIM 606999
- ↑ «Copia arquivada». Consultado em 24 de abril de 2014. Cópia arquivada em 4 de dezembro de 2008
- ↑ Wong LJ, Frey PA (1974). «Galactose-1-phosphate uridylyltransferase: rate studies confirming a uridylyl-enzyme intermediate on the catalytic pathway». Biochemistry. 13 (19): 3889–3894. PMID 4606575. doi:10.1021/bi00716a011
- ↑ «Copia arquivada». Consultado em 24 de abril de 2014. Cópia arquivada em 3 de março de 2016
- ↑ a b c d Wedekind JE, Frey PA, Rayment I (1995). «Three-dimensional structure of galactose-1-phosphate uridylyltransferase from Escherichia coli at 1.8 A resolution». Biochemistry. 34 (35): 11049–61. PMID 7669762. doi:10.1021/bi00035a010
- ↑ a b «Galactose-1-phosphate uridylyltransferase». Consultado em 8 de dezembro de 2011
- ↑ Seyrantepe V, Ozguc M, Coskun T, Ozalp I, Reichardt JK (1999). «Identification of mutations in the galactose-1-phosphate uridyltransferase (GALT) gene in 16 Turkish patients with galactosemia, including a novel mutation of F294Y. Mutation in brief no. 235. Online». Hum. Mutat. 13 (4). 339 páginas. PMID 10220154. doi:10.1002/(SICI)1098-1004(1999)13:4<339::AID-HUMU18>3.0.CO;2-S
- ↑ a b Fridovich-Keil JL (2006). «Galactosemia: the good, the bad, and the unknown». J. Cell. Physiol. 209 (3): 701–5. PMID 17001680. doi:10.1002/jcp.20820
- ↑ a b c d Elsas LJ, Langley S, Paulk EM, Hjelm LN, Dembure PP (1995). «A molecular approach to galactosemia». Eur. J. Pediatr. 154 (7 Suppl 2): S21–7. PMID 7671959. doi:10.1007/BF02143798
- ↑ «Copia arquivada». Consultado em 24 de abril de 2014. Cópia arquivada em 28 de fevereiro de 2006
- ↑ Dobrowolski SF, Banas RA, Suzow JG, Berkley M, Naylor EW (2003). «Analysis of common mutations in the galactose-1-phosphate uridyl transferase gene: new assays to increase the sensitivity and specificity of newborn screening for galactosemia». J Mol Diagn. 5 (1): 42–7. PMC 1907369
 . PMID 12552079
. PMID 12552079
- ↑ Lai K, Elsas LJ, Wierenga KJ (2009). «Galactose toxicity in animals». IUBMB Life. 61 (11): 1063–74. PMC 2788023. PMID 19859980. doi:10.1002/iub.262
- ↑ «Copia arquivada». Consultado em 24 de abril de 2014. Cópia arquivada em 29 de maio de 2013