Genotoxicidade
A genotoxicidade (/ʒɛno/) é a propriedade que alguns agentes químicos têm de causar danos na informação genética de uma célula originando mutações, que podem produzir um cancro. Embora a genotoxicidade seja frequentemente confundida com mutagenicidade, é preciso ter presente que todos os mutagéneos são genotóxicos, mas algumas substâncias genotóxicas não são mutagénicas. A alteração pode ter efeitos diretos ou indiretos sobre o ADN: indução de mutações, ativação de eventos em momentos inapropriados e danos diretos ao ADN que produzem mutações. As alterações permanentes herdáveis podem afetar as células somáticas do organismo ou as células germinais e passar neste segundo caso a futuras gerações.[1]
As células impedem a expressão de mutações genotóxicas por meio da reparação do ADN ou da apoptose (morte celular); contudo, os danos nem sempre podem ser reparados, o que leva à mutagénese. Para fazerem bioensaios de moléculas genotóxicas, os investigadores verificam os danos no ADN em células expostas a substratos tóxicos. Estes danos no ADN podem ser roturas de dupla cadeia ou de cadeia simples, perda da reparação por excisão, ligações cruzadas, sítios lábeis aos álcalis, mutações pontuais e mutações cromossómicas estruturais e numéricas.[2] A alteração da integridade do material genético pode ser causa de cancros. Como consequência, foram desenvolvidas muitas técnicas sofisticadas, como o Teste de Ames, testes toxicológicos in vitro e in vivo, e ensaios Comet para avaliar o potencial que os distintos compostos químicos têm de causar danos no ADN que podem conduzir ao cancro.
Mecanismo
As substâncias genotóxicas induzem danos no material genético nas células por meio de interações com a sequência e estrutura do ADN. Por exemplo, o metal de transição crómio interage com o ADN no seu estado de oxidação de alta valência, incorrendo em lesões de ADN que produzem carcinogénese. Chega-se ao estado de oxidação metaestável Cr(V) pela ativação redutiva. Realizaram-se experiências para estudar a interação entre o ADN e o crómio carcinogénico usando o complexo Cr(V)-Salen no estado de oxidação específico.[3] A interação afeta especificamente o nucleótido guanina (G) na sequência genética. Para facilitar a interação entre o complexo Cr(V)-Salen com a base guanina, os investigadores modificaram as bases a 8-oxo-G para assim ter um local específico de oxidação. A reação entre as duas moléculas causou lesões no ADN; as duas lesões observadas no local da base modificada eram a guanidinohidantoína e a espiroiminodihidantoína. Para realizar uma maior análise da lesão, observou-se que a polimerase parava no local e a adenina era incorporada incorretamente na sequência de ADN no local oposto à base 8-oxo-G. Portanto, estas lesões contêm predominantemente transversões G→T. O crómio de alta valência atua como um carcinogéneo, já que se encontrou que "o mecanismo de danos e produtos da oxidação de bases para a interação entre o crómio de alta valência e o ADN... são relevantes para a formação in vivo de danos no ADN que originam cancro em populações humanas expostas ao cromato".[3] Em consequência, isto mostra como o crómio de alta valência pode atuar como carcinogéneo com xenobióticos que formam 8-oxo-G.[3]
Outro exemplo de substâncias genotóxicas que causam danos no ADN são os alcaloides de pirrolizidina (PAs). Estas substâncias encontram-se principalmente em espécies de plantas e são venenosas para os animais, incluído os humanos; aproximadamente metade deles foram identificados como genotóxicos e muitos como tumorogénicos. Os investigadores concluíram a partir dos testes realizados que quando são ativados metabolicamente, os "PAs produzem adutos do ADN, ligações cruzadas no ADN, roturas nas cadeias de ADN, troca de cromatídeos irmãos, micronúcleos, aberrações cromossómicas, mutações génicas e mutações cromossómicas in vivo e in vitro."[4] As mutações mais comuns nos genes são transversões G:C → T:A e substituições de bases em tandem. Os alcaloides de pirrolizidina são mutagénicos in vivo e in vitro e, portanto, responsáveis pela carcinogénese fundamentalmente no fígado.[4] A planta boraginácea confrei é um exemplo de uma espécie de planta que contém catorze PAs diferentes. Os metabolitos ativos interagem com o ADN causando danos no ADN, induzem a mutação e o desenvolvimento do cancro em células endoteliais do fígado e nos hepatócitos. Descobriu-se no final que o "confrei é mutagénico no fígado, e os PAs contidos no confrei parecem ser responsáveis pela toxicidade induzida pelo confrei e a indução de tumores,".[5]
Técnicas para realizar testes
O propósito dos testes de genotoxicidade é determinar se um substrato influenciará no material genético ou pode causar cancro. Podem ser realizados com bactérias, levedos e células de mamífero.[2] Com o conhecimento obtido nos testes, pode ser controlado o desenvolvimento precoce de organismos vulneráveis a substâncias genotóxicas.[1]
Ensaio de mutação inversa bacteriana
o ensaio de mutação inversa bacteriana, também conhecido como Teste de Ames, é utilizado em laboratórios para fazer testes para as mutações génicas. A técnica usa muitas cepas bacterianas para comparar as diferentes alterações originadas no material genético. O resultado do teste deteta a maioria dos carcinogéneos genotóxicos e alterações genéticas; os tipos de mutações detetados são mutações com mudança da matriz de leitura e as substituições de bases.[6]
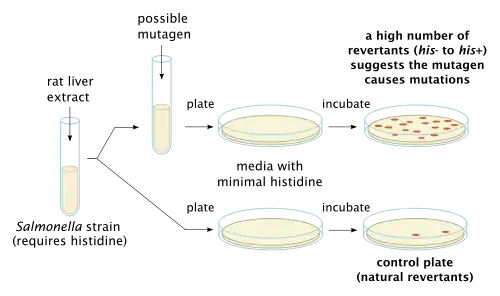
Testes toxicológicos in vitro
O propósito dos testes in vitro é determinar se um substrato, produto, ou fator ambiental induz danos genéticos. Uma técnica utiliza ensaios citogenéticos em diferentes células de mamíferos.[6] Os tipos de anomalias detetadas nas células afetadas por uma substância genotóxica são falhas em cromossomas e cromatídeos, roturas cromossómicas, deleções de cromatídeos, fragmentações, translocações, e rearranjos complexos, entre outras. Os efeitos clastogénicos ou aneugénicos devidos a danos genotóxicos causam um incremento da frequência de aberrações estruturais e numéricas do material genético.[6] Isto é similar ao Teste de micronúcleos e ao ensaio de aberrações cromossómicas, que detetam aberrações cromossómicas estruturais e numéricas em células de mamífero.[1] Num tecido de mamífero determinado, pode-se realizar um ensaio de linfoma TK+/- de rato para testar as alterações no material genético.[6] As mutações génicas são normalmente mutações pontuais que alteram só uma base da sequência genética, alterando o transcrito e a sequência de aminoácidos; estas mutações pontuais podem ser substituições, deleções, mudança da matriz de leitura e rearranjos. Além disso, a integridade dos cromossomas pode ser alterada pela perda de cromossomas e as lesões clastogénicas que causam deleções multilocus e em múltiplos genes. O tipo específico de dano é determinado pelo tamanho das colónias, distinguindo entre mutações genéticas (mutagéneos) e aberrações cromossómicas (clastogénios).[6] O teste do ensaio SOS/umu avalia a capacidade de uma substância de induzir danos no ADN, baseando-se em alterações na indução da resposta SOS devido a danos no ADN. Os benefícios desta técnica são que é um método simples e rápido apropriado para numerosas substâncias. Estas técnicas são realizadas para analisar amostras de água ou águas residuais encontradas no meio ambiente.[7]
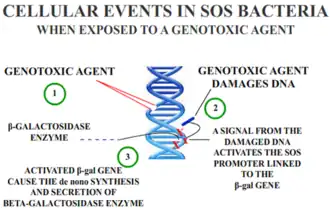
Testes in vivo
O objetivo dos testes in vivo é determinar o potencial de danos no ADN que pode afetar a estrutura cromossómica ou alterar o aparelho mitótico que causa alterações no número de cromossomas; os fatores que poderiam influir na genotoxicidade são a ADME (absorção, distribuição, metabolismo e excreção) e a reparação do ADN. Pode também detetar agentes genotóxicos que passam despercebidos em testes in vitro. O resultado positivo de danos cromossómicos induzidos é um incremento na frequência de PCEs (eritrócitos policromáticos) micronucleados.[6] Um micronúcleo é uma pequena estrutura separada do núcleo que contém ADN nuclear originado de fragmentos de ADN ou de cromossomas completos que não foram incorporados na célula filha durante a mitose. As causas destas estruturas são a perda mitótica de fragmentos de cromossomas acêntricos (clastogenicidade), problemas mecânicos devido a roturas e trocas nos cromossomas, perda mitótica de cromossomas (aneugenicidade), e apoptose. O teste de micronúcleos in vivo é similar ao in vitro porque verifica as aberrações cromossómicas estruturais e numéricas em células de mamíferos, especialmente em células sanguíneas de ratas.[6]
Ensaio Comet
Os ensaios Comet são uns dos testes mais comuns para a genotoxicidade. A técnica implica lisar células usando detergentes e sais. O ADN libertado da célula lisada é submetido a eletroforese em gel de agarose em condições de pH neutro. As células que contêm ADN com um número aumentado de roturas de dupla cadeia migrarão mais rapidamente para o ânodo. Esta técnica é vantajosa porque deteta baixos níveis de danos no ADN, requer somente um número muito pequeno de células, é mais barata que muitas outras técnicas, fácil de executar, e mostra rapidamente os resultados. Contudo, não identifica o mecanismo que subjaz no efeito genotóxico ou o composto ou componente químico exato causador das roturas.[8]
Cancro
Os efeitos genotóxicos como as deleções, roturas e rearranjos podem originar um cancro se os danos não ocasionarem imediatamente a morte celular. Regiões sensíveis à rotura, chamadas sítios frágeis, podem ser o resultado da ação de agentes genotóxicos (como pesticidas). Alguns compostos químicos têm a capacidade de induzir sítios frágeis em regiões do cromossoma onde há oncogenes, o que produz efeitos carcinogénicos. Em consonância com isto, a exposição laboral a algumas misturas de pesticidas está positivamente correlacionada com um incremento de danos genotóxicos nas pessoas expostas. Os danos no ADN não têm uma gravidade uniforme nas populações porque varia a capacidade de cada indivíduo de ativar ou desintoxicar as substâncias genotóxicas, o que origina uma variabilidade na incidência de cancro entre indivíduos. A diferença em capacidade de desintoxicar certos compostos deve-se aos polimorfismos herdados pelos indivíduos de genes que intervêm no metabolismo do composto químico. As diferenças podem também ser atribuídas a variações individuais em eficiência dos mecanismos de reparações do ADN.[9] O metabolismo de alguns compostos químicos causa a produção de espécies reativas do oxigénio, o que é um possível mecanismo de genotoxicidade. Isto pode ser observado no metabolismo do arsénio, que produz radicais hidroxilo, que se sabe serem causadores de efeitos genotóxicos.[10] De forma similar, as espécies reativas do oxigénio foram implicadas na genotoxicidade causada por partículas e fibras. A genotoxicidade de partículas fibrosas e não fibrosas é caracterizada pela alta produção de espécies reativas do oxigénio nas células inflamatórias.[11]
Xenotoxinas implicadas nos quatro tipos de cancro mais comuns no mundo
Foram identificados os principais agentes genotóxicos responsáveis pelos quatro cancros mais comuns em todo o mundo: do pulmão, mama, cólon e estômago. O cancro do pulmão é o cancro mais frequente no mundo, tanto em casos anuais (1,61 milhões de casos; 12,7 % de todos os casos de cancro) como em falecimentos (1,38 milhões de mortes; 18,2 % de todas as mortes por cancro).[12] O fumo do tabaco é a principal causa de cancro pulmonar. Por exemplo, o risco estimado para este cancro indica que o fumo do tabaco é responsável por 90 % dos cancros do pulmão nos Estados Unidos. O fumo do tabaco contém mais de 5300 compostos químicos identificados. Os carcinogéneos mais significativos no fumo do tabaco foram determinados aplicando o conceito de "margem de exposição".[13] Segundo essa abordagem, os compostos tumorogénicos no fumo do tabaco eram, por ordem de importância, a acroleína, formaldeído, acrilonitrilo, 1,3-butadieno, cádmio, acetaldeído, óxido de etileno e isopreno. Em geral, estes compostos são genotóxicos e causam danos no ADN. Como exemplos, foram reportados os efeitos que danificam o ADN da acroleína,[14] o formaldeído,[15] e o acrilonitrilo.[16] O cancro da mama é o segundo cancro mais frequente no mundo por casos anuais (1,38 milhões de casos, 10,9 % de todos os casos de cancro), e está no 5.º lugar como causa de morte (458.000, 6,1 % de todas as mortes por cancro).[12] O risco de cancro da mama está associado com níveis sanguíneos persistentemente altos de estrogénios.[17] O estrogénio provavelmente contribui para a carcinogénese da mama por meio dos seguintes processos; (1) conversão metabólica do estrogénio a carcinogéneos genotóxicos mutagénicos, (2) estimulação do crescimento dos tecidos, e (3) repressão dos enzimas de desintoxicação de fase II que metabolizam espécies reativas do oxigénio genotóxicas, resultando assim num aumento dos danos ao ADN oxidativos.[18][19][20] O principal estrogénio humano, o estradiol, pode metabolizar-se a derivados da quinona que formam adutos do ADN.[21] Estes derivados podem causar a eliminação de bases do eixo fosfodiéster da molécula de ADN (por exemplo, despurinação). Esta eliminação pode ser seguida de uma reparação inexata do sítio apurínico originando uma mutação e finalmente o cancro. O cancro colorretal é o terceiro mais frequente no mundo com 1,23 milhões de casos (9,7 % de todos os casos de cancro), e 608.000 mortes (8,0 % de todas as mortes por cancro).[12] O fumo do tabaco também influi neste cancro. Como exemplo, nos Estados Unidos o fumo do tabaco pode ser o responsável por até 20 % de cancros colorretais.[22] Além disso, há evidências substanciais de que os ácidos biliares são um importante fator genotóxico para o cancro do cólon.[23]
Em particular, o ácido biliar ácido desoxicólico causa a produção de espécies reativas do oxigénio que danificam o ADN em células epiteliais do cólon de humanos e roedores.[23] O cancro do estômago é o quarto tipo de cancro mais comum no mundo com 990.000 casos (7,8% de todos os casos de cancro) e 738.000 óbitos (9,7 % de todas as mortes por cancro).[12] A infeção por Helicobacter pylori é o principal fator causador de cancro do estômago. A inflamação crónica devido a H. pylori, se não for tratada, costuma ser muito duradoura. A infeção por H. pylori de células do epitélio gástrico causa um aumento da produção de espécies reativas do oxigénio genotóxicas.[24][25] As espécies reativas do oxigénio causam danos no ADN que incluem a formação da importante base alterada 8-oxo-2'-desoxiguanosina. Num estudo retrospetivo recente encontrou-se que o uso de sequestradores de ácidos biliares estava associado com uma significativa redução de risco de cancro gástrico, o que sugere que os ácidos biliares podem contribuir para o cancro do estômago.[26]
Quimioterapia genotóxica
A quimioterapia genotóxica é o tratamento do cancro usando um ou mais fármacos genotóxicos. O tratamento faz parte tradicionalmente do regime de quimioterapia estandardizado. Ao utilizar as propriedades destrutivas dos tratamentos com genotoxinas pretende-se induzir danos no ADN nas células cancerosas. Os danos feitos às células do cancro transmitem-se às células cancerosas descendentes à medida que continue a proliferação celular. Se estes danos forem suficientemente graves, induzem que as células sofram apoptose.[27]
Riscos
Uma desvantagem do tratamento é que muitos fármacos genotóxicos são igualmente efetivos contra as células cancerosas e as células normais. A seletividade da ação de um determinado fármaco está baseada na sensibilidade das próprias células. Assim, embora as células cancerosas, que se dividem rapidamente, sejam especialmente sensíveis a muitos tratamentos farmacológicos, frequentemente também afetam o funcionamento das células normais.[27] Outro risco do tratamento é que muitos fármacos, além de serem genotóxicos, são também mutagénicos e citotóxicos, de tal forma que estes fármacos não estão só limitados a produzir danos no ADN. Além disso, alguns destes fármacos que estão pensados para tratar cancros são também eles próprios carcinogéneos, elevando o risco de aparecimento de cancros secundários, como a leucemia.[27]
Diferentes tratamentos
Esta tabela mostra diferentes tratamentos do cancro baseados em genotóxicos juntamente com exemplos.[27]
Rins
Os danos ao ADN genotóxicos nos rins foram implicados na lesão renal crónica e no carcinoma de células renais.[28] Os estudos feitos em pessoas com deficiências genéticas em vias de reparação do ADN revelaram que os seus rins são especialmente vulneráveis aos danos no ADN como as ligações cruzadas no ADN, roturas no ADN e danos que bloqueiam a transcrição.[28]
Referências
- ↑ a b c Kolle, Susanne (1 de junho de 2012). «Genotoxicity and Carcinogenicity». BASF The Chemical Company. Consultado em 16 de março de 2013. Cópia arquivada em 28 de junho de 2013
- ↑ a b «Genotoxicity: Validated Non-animal Alternatives». AltTox.org. 20 de junho de 2011. Consultado em 16 de março de 2013. Cópia arquivada em 20 de abril de 2008
- ↑ a b c Sugden KD, Campo CK, Martin BD (setembro de 2001). «Direct oxidation of guanine and 7,8-dihydro-8-oxoguanine in DNA by a high-valent chromium complex: a possible mechanism for chromate genotoxicity». Chemical Research in Toxicology. 14 (9): 1315–22. PMID 11559048. doi:10.1021/tx010088+
- ↑ a b Chen T, Mei N, Fu PP (abril de 2010). «Genotoxicity of pyrrolizidine alkaloids». Journal of Applied Toxicology. 30 (3): 183–96. PMC 6376482
 . PMID 20112250. doi:10.1002/jat.1504
. PMID 20112250. doi:10.1002/jat.1504
- ↑ Mei N, Guo L, Fu PP, Fuscoe JC, Luan Y, Chen T (outubro de 2010). «Metabolism, genotoxicity, and carcinogenicity of comfrey». Journal of Toxicology and Environmental Health Part B: Critical Reviews. 13 (7–8): 509–26. Bibcode:2010JTEHB..13..509M. PMC 5894094. PMID 21170807. doi:10.1080/10937404.2010.509013
- ↑ a b c d e f g Furman, Grace (17 de abril de 2008). «Genotoxicity Testing for Pharmaceuticals Current and Emerging Practices» (PDF). Paracelsus, Inc. Consultado em 16 de março de 2013. Cópia arquivada (PDF) em 16 de janeiro de 2014
- ↑ Končar, Helena (2011). «In Vitro Genotoxicity Testing». National Institute of Biology. Consultado em 16 de março de 2013. Cópia arquivada em 7 de março de 2013
- ↑ Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, et al. (2000). «Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing» (PDF). Environmental and Molecular Mutagenesis. 35 (3): 206–21. Bibcode:2000EnvMM..35..206T. PMID 10737956. doi:10.1002/(SICI)1098-2280(2000)35:3<206::AID-EM8>3.0.CO;2-J. Consultado em 20 de março de 2025. Cópia arquivada (PDF) em 24 de dezembro de 2012
- ↑ Bolognesi, Claudia (junho de 2003). «Genotoxicity of pesticides: A review of human biomonitoring studies». Mutation Research. 543 (3): 251–272. Bibcode:2003MRRMR.543..251B. PMID 12787816. doi:10.1016/S1383-5742(03)00015-2
- ↑ Liu SX, Athar M, Lippai I, Waldren C, Hei TK (fevereiro de 2001). «Induction of oxyradicals by arsenic: implication for mechanism of genotoxicity». Proceedings of the National Academy of Sciences of the United States of America. 98 (4): 1643–8. Bibcode:2001PNAS...98.1643L. PMC 29310. PMID 11172004. doi:10.1073/pnas.98.4.1643
- ↑ Schins RP (janeiro de 2002). «Mechanisms of genotoxicity of particles and fibers». Inhalation Toxicology. 14 (1): 57–78. Bibcode:2002InhTx..14...57S. PMID 12122560. doi:10.1080/089583701753338631
- ↑ a b c d Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM (dezembro de 2010). «Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008». Int J Cancer. 127 (12): 2893–2917. PMID 21351269. doi:10.1002/ijc.25516
- ↑ Cunningham FH, Fiebelkorn S, Johnson M, Meredith C (novembro de 2011). «A novel application of the Margin of Exposure approach: segregation of tobacco smoke toxicants». Food Chem Toxicol. 49 (11): 2921–33. PMID 21802474. doi:10.1016/j.fct.2011.07.019
- ↑ Liu D, Cheng Y, Tang Z, Mei X, Cao X, Liu J (janeiro de 2022). «Toxicity mechanism of acrolein on DNA damage and apoptosis in BEAS-2B cells: Insights from cell biology and molecular docking analyses». Toxicology. 466. 153083 páginas. Bibcode:2022Toxgy.46653083L. PMID 34958888. doi:10.1016/j.tox.2021.153083
- ↑ Mulderrig L, Garaycoechea JI, Tuong ZK, Millington CL, Dingler FA, Ferdinand JR, Gaul L, Tadross JA, Arends MJ, O'Rahilly S, Crossan GP, Clatworthy MR, Patel KJ (dezembro de 2021). «Aldehyde-driven transcriptional stress triggers an anorexic DNA damage response». Nature. 600 (7887): 158–163. Bibcode:2021Natur.600..158M. PMID 34819667. doi:10.1038/s41586-021-04133-7. hdl:20.500.11820/5c198932-e822-43cb-a2cf-56369f454de9
- ↑ Pu X, Kamendulis LM, Klaunig JE (setembro de 2009). «Acrylonitrile-induced oxidative stress and oxidative DNA damage in male Sprague-Dawley rats». Toxicol Sci. 111 (1): 64–71. PMC 2726299. PMID 19546159. doi:10.1093/toxsci/kfp133
- ↑ Yager JD, Davidson NE (janeiro de 2006). «Estrogen carcinogenesis in breast cancer». N Engl J Med. 354 (3): 270–82. PMID 16421368. doi:10.1056/NEJMra050776
- ↑ Ansell PJ, Espinosa-Nicholas C, Curran EM, Judy BM, Philips BJ, Hannink M, Lubahn DB (janeiro de 2004). «In vitro and in vivo regulation of antioxidant response element-dependent gene expression by estrogens». Endocrinology. 145 (1): 311–7. PMID 14551226. doi:10.1210/en.2003-0817
- ↑ Belous AR, Hachey DL, Dawling S, Roodi N, Parl FF (janeiro de 2007). «Cytochrome P450 1B1-mediated estrogen metabolism results in estrogen-deoxyribonucleoside adduct formation». Cancer Res. 67 (2): 812–7. PMID 17234793. doi:10.1158/0008-5472.CAN-06-2133
- ↑ Bolton JL, Thatcher GR (janeiro de 2008). «Potential mechanisms of estrogen quinone carcinogenesis». Chem Res Toxicol. 21 (1): 93–101. PMC 2556295. PMID 18052105. doi:10.1021/tx700191p
- ↑ Yue W, Santen RJ, Wang JP, Li Y, Verderame MF, Bocchinfuso WP, Korach KS, Devanesan P, Todorovic R, Rogan EG, Cavalieri EL (setembro de 2003). «Genotoxic metabolites of estradiol in breast: potential mechanism of estradiol induced carcinogenesis». J Steroid Biochem Mol Biol. 86 (3–5): 477–86. PMID 14623547. doi:10.1016/s0960-0760(03)00377-7
- ↑ Giovannucci E, Martínez ME (dezembro de 1996). «Tobacco, colorectal cancer, and adenomas: a review of the evidence». J Natl Cancer Inst. 88 (23): 1717–30. PMID 8944002. doi:10.1093/jnci/88.23.1717
- ↑ a b Bernstein H, Bernstein C (janeiro de 2023). «Bile acids as carcinogens in the colon and at other sites in the gastrointestinal system». Exp Biol Med (Maywood). 248 (1): 79–89. PMC 9989147. PMID 36408538. doi:10.1177/15353702221131858
- ↑ Ding SZ, Minohara Y, Fan XJ, Wang J, Reyes VE, Patel J, Dirden-Kramer B, Boldogh I, Ernst PB, Crowe SE (agosto de 2007). «Helicobacter pylori infection induces oxidative stress and programmed cell death in human gastric epithelial cells». Infect Immun. 75 (8): 4030–9. PMC 1952011. PMID 17562777. doi:10.1128/IAI.00172-07
- ↑ Handa O, Naito Y, Yoshikawa T (2011). «Redox biology and gastric carcinogenesis: the role of Helicobacter pylori». Redox Rep. 16 (1): 1–7. PMC 6837368. PMID 21605492. doi:10.1179/174329211X12968219310756
- ↑ Noto JM, Piazuelo MB, Shah SC, Romero-Gallo J, Hart JL, Di C, Carmichael JD, Delgado AG, Halvorson AE, Greevy RA, Wroblewski LE, Sharma A, Newton AB, Allaman MM, Wilson KT, Washington MK, Calcutt MW, Schey KL, Cummings BP, Flynn CR, Zackular JP, Peek RM (maio de 2022). «Iron deficiency linked to altered bile acid metabolism promotes Helicobacter pylori-induced inflammation-driven gastric carcinogenesis». J Clin Invest. 132 (10). PMC 9106351. PMID 35316215. doi:10.1172/JCI147822
- ↑ a b c d Walsh, Declan (18 de novembro de 2011). «Genotoxic Drugs». CancerQuest. Consultado em 16 de março de 2013. Cópia arquivada em 2 de março de 2013
- ↑ a b Garaycoechea JI, Quinlan C, Luijsterburg MS (abril de 2023). «Pathological consequences of DNA damage in the kidney». Nat Rev Nephrol. 19 (4): 229–243. PMID 36702905. doi:10.1038/s41581-022-00671-z
Ver também
- Mutagênicos
- Teratogênese
- Carcinógeno
- Bioindicador
- Epidemiologia genética