Biogênese mitocondrial
A biogênese mitocondrial é o processo pelo qual as células aumentam o número de mitocôndrias.[1][2] Foi descrito pela primeira vez por John Holloszy na década de 1960, quando foi descoberto que o treinamento de resistência física induzia níveis mais elevados de conteúdo mitocondrial, levando a uma maior captação de glicose pelos músculos.[3] A biogênese mitocondrial é ativada por vários sinais diferentes durante períodos de estresse celular ou em resposta a estímulos ambientais, como exercícios aeróbicos.[1][2][4]
Visão geral
A capacidade de uma mitocôndria de se autorreplicar está enraizada em sua história evolutiva. É comumente pensado que as mitocôndrias descendem de células que formaram relações endossimbióticas com α-protobactérias; elas têm seu próprio genoma para replicação.[5] No entanto, evidências recentes sugerem que as mitocôndrias podem ter evoluído sem simbiose.[6] A mitocôndria é um regulador chave da atividade metabólica da célula e também é uma organela importante tanto na produção quanto na degradação de radicais livres.[7] Postula-se que um maior número de cópias mitocondriais (ou maior massa mitocondrial) é protetor para a célula.[8]
As mitocôndrias são produzidas a partir da transcrição e tradução de genes tanto no genoma nuclear quanto no genoma mitocondrial. A maior parte da proteína mitocondrial vem do genoma nuclear, enquanto o genoma mitocondrial codifica partes da cadeia de transporte de elétrons junto com o rRNA e o tRNA mitocondriais. A biogênese mitocondrial aumenta as enzimas metabólicas para glicólise, fosforilação oxidativa e, finalmente, uma maior capacidade metabólica mitocondrial. Entretanto, dependendo dos substratos energéticos disponíveis e do estado redox da célula, a célula pode aumentar ou diminuir o número e o tamanho das mitocôndrias.[9] De modo crítico, o número e a morfologia mitocondrial variam de acordo com o tipo de célula e a demanda específica do contexto, de modo que o equilíbrio entre a fusão/fissão mitocondrial regula a distribuição, a morfologia e a função mitocondrial.[10][9]
Importação de proteínas
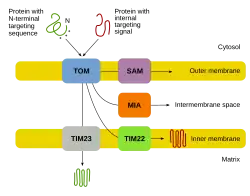
Como a maior parte da proteína mitocondrial vem do genoma nuclear, as proteínas precisam ser adequadamente direcionadas e transportadas para as mitocôndrias para desempenhar suas funções.[9][11][12] Primeiro, o mRNA é traduzido no citosol da célula.[11][12] As proteínas precursoras desdobradas resultantes poderão então atingir seus respectivos compartimentos mitocondriais.[12][11] As proteínas precursoras serão transportadas para uma das quatro áreas da mitocôndria, que incluem a membrana externa, a membrana interna, o espaço intermembranar e a matriz.[11][12] Todas as proteínas entrarão na mitocôndria por uma translocase na membrana mitocondrial externa.[12][11][5] Algumas proteínas terão um sinal de direcionamento N-terminal, e essas proteínas serão detectadas e transportadas para a matriz, onde serão clivadas e dobradas.[13][12][11] Outras proteínas podem ter informações de direcionamento em suas sequências e não incluirão um sinal N-terminal.[12][11] Durante as últimas duas décadas, os pesquisadores descobriram mais de trinta proteínas que participam da importação de proteínas mitocondriais.[12] À medida que os pesquisadores aprendem mais sobre essas proteínas e como elas chegam aos respectivos compartimentos mitocondriais que as utilizam, torna-se evidente que há uma infinidade de processos que trabalham juntos na célula para permitir a biogênese mitocondrial.[12][9]
Fusão e fissão
As mitocôndrias são altamente versáteis e são capazes de mudar sua forma por meio de eventos de fissão e fusão.[10][9] Definitivamente, a fissão é o evento de uma única entidade se separando, enquanto a fusão é o evento de duas ou mais entidades se unindo para formar um todo.[9] Os processos de fissão e fusão se opõem e permitem que a rede mitocondrial se remodele constantemente.[10][9] Se um estímulo induz uma mudança no equilíbrio de fissão e fusão numa célula, pode alterar significativamente a rede mitocondrial.[10][14] Por exemplo, um aumento na fissão mitocondrial criaria muitas mitocôndrias fragmentadas, o que se demonstrou ser útil para eliminar mitocôndrias danificadas e para criar mitocôndrias menores para transporte eficiente para áreas com maior demanda de energia.[14][15] Portanto, alcançar um equilíbrio entre esses mecanismos permite que uma célula tenha a organização adequada de sua rede mitocondrial durante a biogênese e pode ter um papel importante na adaptação muscular ao estresse fisiológico.[14]
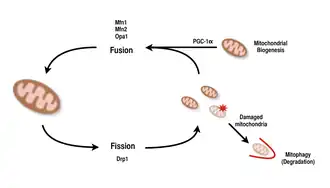
Em mamíferos, a fusão e a fissão mitocondrial são controladas por GTPases da família da dinamina.[9][14] O processo de fissão mitocondrial é dirigido por Drp1, um membro da família da dinamina citosólica.[9][10] Esta proteína forma uma espiral ao redor das mitocôndrias e se contrai para separar as membranas externa e interna da organela.[15] Por outro lado, o processo de fusão é dirigido por diferentes proteínas dinaminas ancoradas à membrana em diferentes níveis das mitocôndrias.[14] A fusão ao nível da membrana mitocondrial externa é mediada por Mfn1 e Mfn2 (mitofusinas 1 e 2),[16] e a fusão ao nível da membrana mitocondrial interna é mediada por Opa1.[9][13][14] Vários estudos de pesquisa observaram aumentos correlacionados entre a capacidade respiratória mitocondrial com a expressão dos genes Mfn1, Mnf2 e Drp1 após exercícios de resistência.[15][16] Portanto, é sustentado que a reorganização da rede mitocondrial nas células musculares desempenha um papel importante na resposta ao exercício.[4][14][16]
Regulação
PGC-1α, um membro da família de coativadores transcricionais do receptor gama proliferador de peroxissoma (PGC), é o principal regulador da biogênese mitocondrial.[1][2][17] É conhecido por coativar o fator respiratório nuclear 2 (NRF2/GABPA) e, juntamente com o NRF-2, coativar o fator respiratório nuclear 1 (NRF1).[16][17] Os NRFs, por sua vez, ativam o fator de transcrição mitocondrial A (tfam), que é diretamente responsável pela transcrição de proteínas mitocondriais codificadas no núcleo.[16][17] Isso inclui proteínas mitocondriais estruturais, bem como aquelas envolvidas na transcrição, tradução e reparo do mtDNA.[17] PGC-1β, uma proteína estruturalmente semelhante à PGC-1α, também está envolvida na regulação da biogênese mitocondrial, mas difere por não aumentar em resposta ao exercício.[5][18][17] Embora tenham ocorrido aumentos significativos nas mitocôndrias encontradas em tecidos onde o PGC-1α é superexpresso, à medida que o cofator interage com esses fatores de transcrição essenciais, os camundongos knockout com PGC-1α interrompido ainda são viáveis e apresentam abundância mitocondrial normal.[18][5][17] Assim, o PGC-1α não é necessário para o desenvolvimento normal das mitocôndrias em ratos, mas quando submetidos a estresse fisiológico, esses ratos apresentam tolerância diminuída em comparação com ratos com níveis normais de PGC-1α.[5][17][18] Da mesma forma, em ratos knockout com PGC-1β interrompido, os ratos apresentaram níveis principalmente normais de função mitocondrial com capacidade diminuída de adaptação ao estresse fisiológico.[19][5] No entanto, um experimento de dupla eliminação de PGC-1α/β criou ratos que morreram principalmente em 24 horas devido a defeitos na maturação mitocondrial do tecido cardíaco.[20] Essas descobertas sugerem que, embora PGC-1α e PGC-1β não estabeleçam individualmente a capacidade de uma célula de realizar a biogênese mitocondrial, juntos eles são capazes de se complementar para a maturação e função mitocondrial ideais durante períodos de estresse fisiológico.[20][5][18]
A cinase ativada por AMP (AMPK) também regula a biogênese mitocondrial por meio da fosforilação e ativação de PGC-1α ao detectar uma deficiência de energia no músculo.[5][17] Em ratos com proporções ATP/AMP reduzidas que ocorreriam durante o exercício, a depleção de energia demonstrou estar correlacionada com a ativação de AMPK.[5][19][17] A ativação de AMPK continuou a ativar PGC-1α e NRFs nesses camundongos, e a biogênese mitocondrial foi estimulada.[5][19][17]
Envelhecimento
Foi demonstrado que a capacidade de biogênese mitocondrial diminui com a idade, e essa diminuição da função mitocondrial tem sido associada ao diabetes e às doenças cardiovasculares.[21][22][23] O envelhecimento e a doença podem induzir alterações nos níveis de expressão de proteínas envolvidas nos mecanismos de fissão e fusão das mitocôndrias, criando assim mitocôndrias disfuncionais.[24][25] Uma hipótese para os resultados prejudiciais do envelhecimento está associada à perda de telômeros, os segmentos finais dos cromossomas que protegem a informação genética da degradação.[22][25] A perda de telômeros também foi associada à diminuição da função mitocondrial.[25][22] A deficiência da telomerase transcriptase reversa (TERT), uma enzima que desempenha um papel na preservação dos telômeros, foi correlacionada com o p53 ativado, uma proteína que suprime o PGC-1α.[25][24][22] Portanto, a perda de telômeros e TERT que ocorre com o envelhecimento tem sido associada à biogênese mitocondrial prejudicada.[22][24][25] A expressão de AMPK também demonstrou diminuir com a idade, o que também pode contribuir para a supressão da biogênese mitocondrial.[5][25]
Aplicações clínicas do direcionamento da biogênese mitocondrial
A biogênese mitocondrial pode ser direcionada para prevenir a proliferação do câncer. Especificamente, dois reguladores de biogênese — PGC1α e c-Myc — podem ser direcionados para prevenir a proliferação do câncer. PGC1α é um componente chave na biogênese mitocondrial - como um coativador transcricional, ele tem como alvo múltiplos fatores de transcrição e o receptor alfa relacionado ao estrogênio (ERRα).[26] Compostos que têm como alvo a via entre PGC1α e ERRα, como o agonista inverso de ERRα, XCT-790, demonstraram diminuir significativamente a biogênese mitocondrial, reduzindo significativamente a proliferação de células cancerígenas e aumentando sua sensibilidade aos agentes quimioterápicos.[27] c-Myc, um fator de transcrição, pode ser inibido durante sua dimerização com a proteína Max por moléculas como IIA6B17[28] e omomyc.[29] A inibição do complexo c-Myc-Max pode bloquear o ciclo celular e induzir apoptose em células cancerígenas.[30]
Notas
- Este artigo foi inicialmente traduzido, total ou parcialmente, do artigo da Wikipédia em inglês cujo título é «Mitochondrial biogenesis».
Referências
- ↑ a b c Valero T (2014). «Editorial (Thematic Issue: Mitochondrial Biogenesis: Pharmacological Approaches)». Current Pharmaceutical Design. 20 (35): 5507–5509. PMID 24606795. doi:10.2174/138161282035140911142118
- ↑ a b c Sanchis-Gomar F, García-Giménez JL, Gómez-Cabrera MC, Pallardó FV (2014). «Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches». Current Pharmaceutical Design. 20 (35): 5619–33. PMID 24606801. doi:10.2174/1381612820666140306095106
- ↑ Holloszy JO (abril de 2011). «RRegulation of Mitochondrial Biogenesis and GLUT4 Expression by Exercise». Comprehensive Physiology. 1 (2): 921–40. ISBN 9780470650714. PMID 23737207. doi:10.1002/cphy.c100052
- ↑ a b Boushel R, Lundby C, Qvortrup K, Sahlin K (outubro de 2014). «Mitochondrial plasticity with exercise training and extreme environments». Exercise and Sport Sciences Reviews. 42 (4): 169–74. PMID 25062000. doi:10.1249/JES.0000000000000025
- ↑ a b c d e f g h i j k Jornayvaz FR, Shulman GI (2010). «Regulation of mitochondrial biogenesis». Essays in Biochemistry. 47: 69–84. PMC 3883043. PMID 20533901. doi:10.1042/bse0470069
- ↑ Harish A, Kurland CG (dezembro de 2017). «Mitochondria are not captive bacteria». Journal of Theoretical Biology. 434: 88–98. Bibcode:2017JThBi.434...88H. PMID 28754286. doi:10.1016/j.jtbi.2017.07.011
- ↑ Bevilacqua L, Ramsey JJ, Hagopian K, Weindruch R, Harper ME (maio de 2004). «Effects of short- and medium-term calorie restriction on muscle mitochondrial proton leak and reactive oxygen species production». American Journal of Physiology. Endocrinology and Metabolism. 286 (5): E852-61. PMID 14736705. doi:10.1152/ajpendo.00367.2003
- ↑ Takahashi, Paul Y.; Jenkins, Gregory D.; Welkie, Benjamin P.; McDonnell, Shannon K.; Evans, Jared M.; Cerhan, James R.; Olson, Janet E.; Thibodeau, Steven N.; Cicek, Mine S. (25 de outubro de 2018). «Association of mitochondrial DNA copy number with self-rated health status». The Application of Clinical Genetics (em inglês): 121–127. doi:10.2147/TACG.S167640. Consultado em 16 de julho de 2025
- ↑ a b c d e f g h i j Mishra P, Chan DC (fevereiro de 2016). «Metabolic regulation of mitochondrial dynamics». The Journal of Cell Biology. 212 (4): 379–87. PMC 4754720. PMID 26858267. doi:10.1083/jcb.201511036
- ↑ a b c d e Bertholet AM, Delerue T, Millet AM, Moulis MF, David C, Daloyau M, et al. (junho de 2016). «Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity». Neurobiology of Disease. 90: 3–19. PMID 26494254. doi:10.1016/j.nbd.2015.10.011
- ↑ a b c d e f g Dudek J, Rehling P, van der Laan M (fevereiro de 2013). «Mitochondrial protein import: common principles and physiological networks». Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1833 (2): 274–85. PMID 22683763. doi:10.1016/j.bbamcr.2012.05.028
- ↑ a b c d e f g h i Baker MJ, Frazier AE, Gulbis JM, Ryan MT (setembro de 2007). «Mitochondrial protein-import machinery: correlating structure with function». Trends in Cell Biology. 17 (9): 456–64. PMID 17825565. doi:10.1016/j.tcb.2007.07.010
- ↑ a b Ventura-Clapier R, Garnier A, Veksler V (julho de 2008). «Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha». Cardiovascular Research. 79 (2): 208–17. PMID 18430751. doi:10.1093/cvr/cvn098
- ↑ a b c d e f g Youle RJ, van der Bliek AM (agosto de 2012). «Mitochondrial fission, fusion, and stress». Science. 337 (6098): 1062–5. Bibcode:2012Sci...337.1062Y. PMC 4762028. PMID 22936770. doi:10.1126/science.1219855
- ↑ a b c Bo H, Zhang Y, Ji LL (julho de 2010). «Redefining the role of mitochondria in exercise: a dynamic remodeling». Annals of the New York Academy of Sciences. 1201 (1): 121–8. Bibcode:2010NYASA1201..121B. PMID 20649548. doi:10.1111/j.1749-6632.2010.05618.x
- ↑ a b c d e Cartoni R, Léger B, Hock MB, Praz M, Crettenand A, Pich S, et al. (agosto de 2005). «Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise». The Journal of Physiology. 567 (Pt 1): 349–58. PMC 1474174. PMID 15961417. doi:10.1113/jphysiol.2005.092031
- ↑ a b c d e f g h i j Johri A, Chandra A, Flint Beal M (setembro de 2013). «PGC-1α, mitochondrial dysfunction, and Huntington's disease». Free Radical Biology & Medicine. 62: 37–46. PMC 3722269. PMID 23602910. doi:10.1016/j.freeradbiomed.2013.04.016
- ↑ a b c d Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, et al. (outubro de 2004). «Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice». Cell. 119 (1): 121–35. PMID 15454086. doi:10.1016/j.cell.2004.09.013
- ↑ a b c Scarpulla RC (julho de 2011). «Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network». Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1813 (7): 1269–78. PMC 3035754. PMID 20933024. doi:10.1016/j.bbamcr.2010.09.019
- ↑ a b Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, et al. (julho de 2008). «Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart». Genes & Development. 22 (14): 1948–61. PMC 2492740. PMID 18628400. doi:10.1101/gad.1661708
- ↑ Handy DE, Loscalzo J (junho de 2012). «Redox regulation of mitochondrial function». Antioxidants & Redox Signaling. 16 (11): 1323–67. PMC 3324814. PMID 22146081. doi:10.1089/ars.2011.4123
- ↑ a b c d e David R (abril de 2011). «Ageing: Mitochondria and telomeres come together». Nature Reviews. Molecular Cell Biology. 12 (4). 204 páginas. PMID 21407239. doi:10.1038/nrm3082
- ↑ Hagen TM, Wehr CM, Ames BN (novembro de 1998). «Mitochondrial decay in aging. Reversal through supplementation of acetyl-L-carnitine and N-tert-butyl-alpha-phenyl-nitrone». Annals of the New York Academy of Sciences. 854 (1): 214–23. Bibcode:1998NYASA.854..214H. PMID 9928432. doi:10.1111/j.1749-6632.1998.tb09904.x
- ↑ a b c Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, et al. (fevereiro de 2011). «Telomere dysfunction induces metabolic and mitochondrial compromise». Nature. 470 (7334): 359–65. Bibcode:2011Natur.470..359S. PMC 3741661. PMID 21307849. doi:10.1038/nature09787
- ↑ a b c d e f Sahin E, DePinho RA (maio de 2012). «Axis of ageing: telomeres, p53 and mitochondria». Nature Reviews. Molecular Cell Biology. 13 (6): 397–404. PMC 3718675. PMID 22588366. doi:10.1038/nrm3352
- ↑ Scarpulla, Richard (2011). «Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network». Biochim Biophys Acta. 1813 (7): 1269–1278. PMC 3035754. PMID 20933024. doi:10.1016/j.bbamcr.2010.09.019
- ↑ Kokabu, Tetsuya; Mori, Taisuke; Matsushima, Hiroshi; Yoriki, Kaori; Kataoka, Hisashi; Tarumi, Yosuke; Kitawaki, Jo (2019). «Antitumor effect of XCT790, an ERRα inverse agonist, on ERα-negative endometrial cancer cells». Cell Oncol (Dordr). 42 (2): 223–235. PMID 30706380. doi:10.1007/s13402-019-00423-5
- ↑ Lu, X.; Vogt, P. K.; Boger, D. L.; Lunec, J. (2008). «Disruption of the MYC transcriptional function by a small-molecule antagonist of MYC/MAX dimerization». Oncology Reports. 19 (3): 825–830. PMID 18288422
- ↑ Demma, Mark; Mapelli, Claudio; Sun, Angie; Bodea, Smaranda; Ruprecht, Benjamin; Javaid, Sarah; Wiswell, Derek; Muise, Eric; Chen, Shiyang (2019). «Omomyc Reveals New Mechanisms To Inhibit the MYC Oncogene». Mol Cell Biol. 39 (22): e00248-19. PMC 6817756. PMID 31501275. doi:10.1128/MCB.00248-19
- ↑ Zhang, Meng; Fan, Hai-Yan; Li, Sheng-Chao (1 de julho de 2015). «Inhibition of c-Myc by 10058-F4 induces growth arrest and chemosensitivity in pancreatic ductal adenocarcinoma». Biomedicine & Pharmacotherapy: 123–128. ISSN 0753-3322. doi:10.1016/j.biopha.2015.05.019. Consultado em 16 de julho de 2025
Bibliografia
- Smith JA, Stallons LJ, Collier JB, Chavin KD, Schnellmann RG (fevereiro de 2015). «Suppression of mitochondrial biogenesis through toll-like receptor 4-dependent mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling in endotoxin-induced acute kidney injury». The Journal of Pharmacology and Experimental Therapeutics. 352 (2): 346–57. PMC 4293437. PMID 25503387. doi:10.1124/jpet.114.221085
- Cameron RB, Beeson CC, Schnellmann RG (dezembro de 2016). «Development of Therapeutics That Induce Mitochondrial Biogenesis for the Treatment of Acute and Chronic Degenerative Diseases». Journal of Medicinal Chemistry. 59 (23): 10411–10434. PMC 5564430. PMID 27560192. doi:10.1021/acs.jmedchem.6b00669
- Whitaker RM, Corum D, Beeson CC, Schnellmann RG (2016). «Mitochondrial Biogenesis as a Pharmacological Target: A New Approach to Acute and Chronic Diseases». Annual Review of Pharmacology and Toxicology. 56: 229–49. PMID 26566156. doi:10.1146/annurev-pharmtox-010715-103155